
We are really excited to share our @biorxivpreprint on "In silico saturation mutagenesis of cancer genes". Here’s a #tweetorial to briefly summarize our findings doi.org/10.1101/2020.0…
Tumors are known to follow an evolutionary process whereby somatic variants can contribute to cancer development. The study of the traces of positive selection of tumorigenic mutations at the level of genes has yielded a compendium of cancer driver genes (intogen.org) 
But not all mutations reported in cancer genes are tumorigenic. Our knowledge of how selection acts at the level of individual mutations is still limited, so we aimed to explore ways for identifying all potential driver mutations in driver genes, even if we did not observe them.

This is not a new endeavour in cancer genomics. Saturation mutagenesis (aka saturation genome editing) has been attempted for a handful of cancer genes in specific experimental systems. See for example pnas.org/content/100/14…, cell.com/ajhg/fulltext/… and nature.com/articles/s4158…
Despite its value, experimental saturation mutagenesis is costly and laborious, thus limited to a handful of cancer genes and experimental systems so far, and it does not necessarily represent the true evolutionary constraints operative across different somatic tissues.
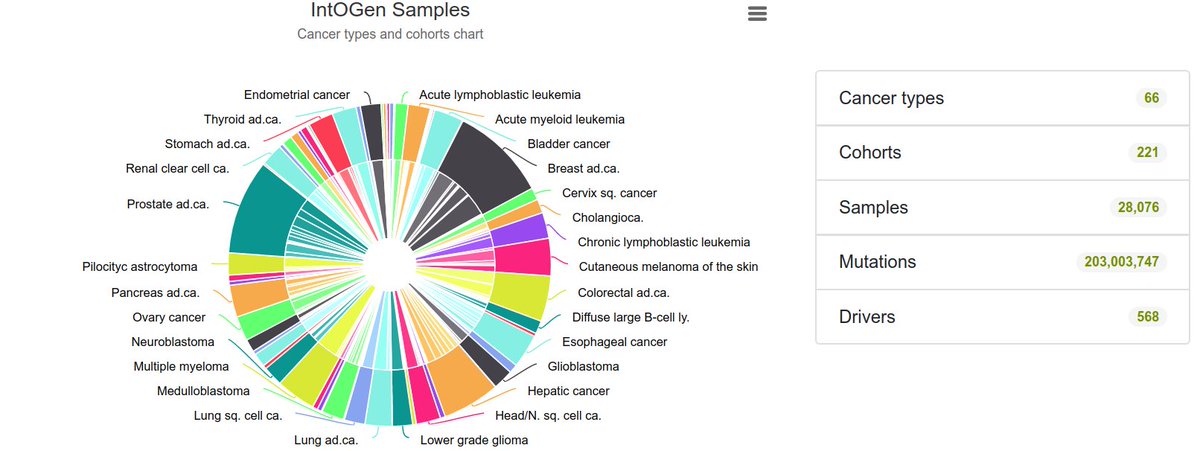
We reasoned that it might be possible to come up with a saturation mutagenesis “in silico” by learning from the large catalogue of mutations in cancer genes across 28,000 tumors comprising 66 cancer types collected by intogen.org
We assembled a training dataset with mutational features computed by intOGen-pipeline for each cancer gene/tumor type, including clusters/hotspots, domains enriched by mutations, biases towards mutation consequences, plus others such as post-translational modif. and conservation

Then we devised a classification method based on gradient boosting (boostDM) trained to distinguish between observed mutations matching a high-excess genomic environment (dNdScv) and random mutations, in a gene and tumor type specific manner.

With our approach we can cast predictions for every single mutation, but we can also leverage the gradient boosting models to bring in SHAP explanations, thereby breaking down forecasts of individual mutations into feature importance weights.

The method showed a remarkable cross-validation accuracy for 105 cancer gene and tumor type specific models. Accuracy was better for genes with a higher number of observed mutations, which is good news, as it suggests that better models are possible given more data.

We validated our approach by comparing our predictions to several independent datasets from experimentally validated and manually curated collections of somatic pathogenic variants. Our approach showed a high overall agreement with a somewhat conservative edge.


Our models have the flexibility to cast quite divergent predictions across tumor types, even within the same gene. EGFR in lung adenocarcinoma and glioblastoma is a prominent example.

This is a reflection of the pervasive diversity of patterns learnt at different cancer genes and tumor type contexts, which can be revealed by the positional distribution of potential drivers as well as by their respective explanations.


The ratio of potential drivers over all possible mutants was lower in oncogenes than in tumor suppressor genes. However, we did not find differences in the ratio of observed over potential drivers.

While the former may be determined by the availability of gain/loss-of-function sites, the latter is shaped by mutation probability and selection.
To better understand the interplay between mutagenesis and selection, we tested whether trinucleotide mutation rates tended to be higher at observed vs non-observed mutations across all potential drivers. We found a positive bias, higher in tumor-suppressor genes.

When considering all observed vs non-observed mutations in driver and non-driver genes, we found this propensity to be higher in non-driver genes, consistently with selection bottlenecks being operative in driver genes and not in non-drivers.

Mutation probability arises as a superposition of different mutational processes. By doing signature fitting we decomposed the sample-wise mutational processes contributing to each potential driver observed.

In doing so we could also assess which mutational processes were dominant at generating each potential driver in each tumor type context. Interestingly, some driver mutations frequently observed across tumor types were best explained by different mutational processes.

Finally, we set out to explore why there are yet-unobserved potential drivers. Would they be observed if more samples were sequenced? False positives of our approach? Negative selection? Do they hold tumorigenic potential which is outcompeted by other frequent mutants?
To address this point we assessed the rate at which yet-unobserved potential drivers would be uncovered if more samples were sequenced, based on random subsampling of sequenced samples, from where we derived a how-close-to-saturation mutational discovery index.

We found that this index across cancer genes is negatively associated with the number of potential driver mutations they bear. Interestingly, cancer genes with weak probability bias tended to be closer to saturation, irrespective of the observed vs potential drivers ratio.

We have shown that in silico saturation mutagenesis is a useful proof-of-concept to look closely into the mechanisms of tumorigenesis with good potential for improvement as new sequenced tumours are available.
The output of the in silico saturation mutagenesis for 120 high-quality models alongside the forecasts for all mutations observed in intogen.org are available here: intogen.org/boostdm/search. The source code of our pipeline is in this repo: bitbucket.org/bbglab/boostdm
This is a team effort involving many people. Full credit to co-authors @fran_mj88, @oriol_pich, Abel Gonzalez-Perez and @nlbigas for their huge commitment to this project. Thanks also to @LorisMularoni, Iker Reyes-Salazar, @ETapanari and @clarnedo for their support.
Last but not least, this kind of analysis would have never been possible without the generosity of donors, clinicians and institutional efforts to collect and sequence tumor samples. Shout-out to @cbioportal, @HartwigMedical, @icgc_dcc, #PCAWG, @StJude and #TCGA

