Keep Current with Eric Fauman (he/him)

Stay in touch and get notified when new unrolls are available from this author!
This Thread may be Removed Anytime!
Twitter may remove this content at anytime! Save it as PDF for later use!

@threadreaderapp unroll
Practice here first or read more on our help page!


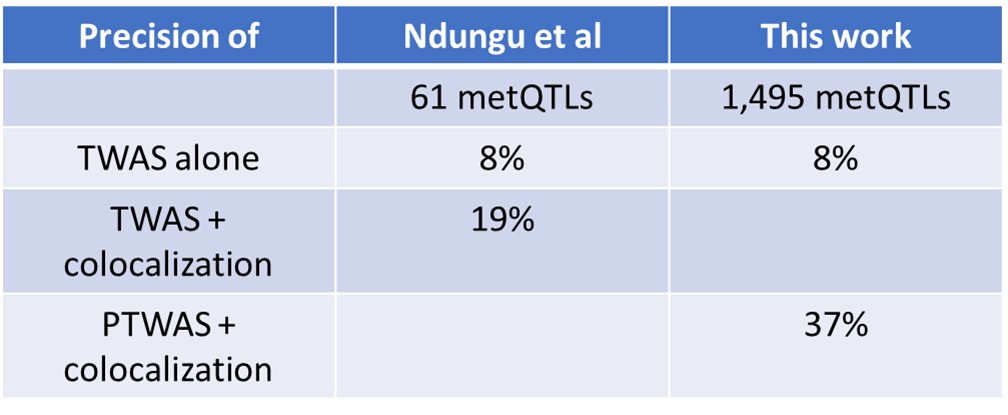


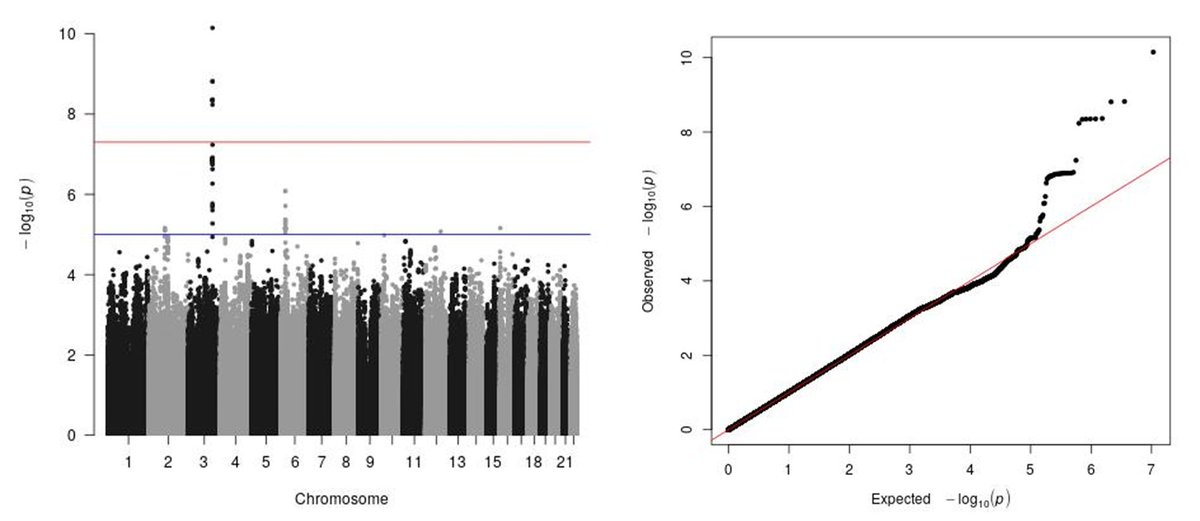





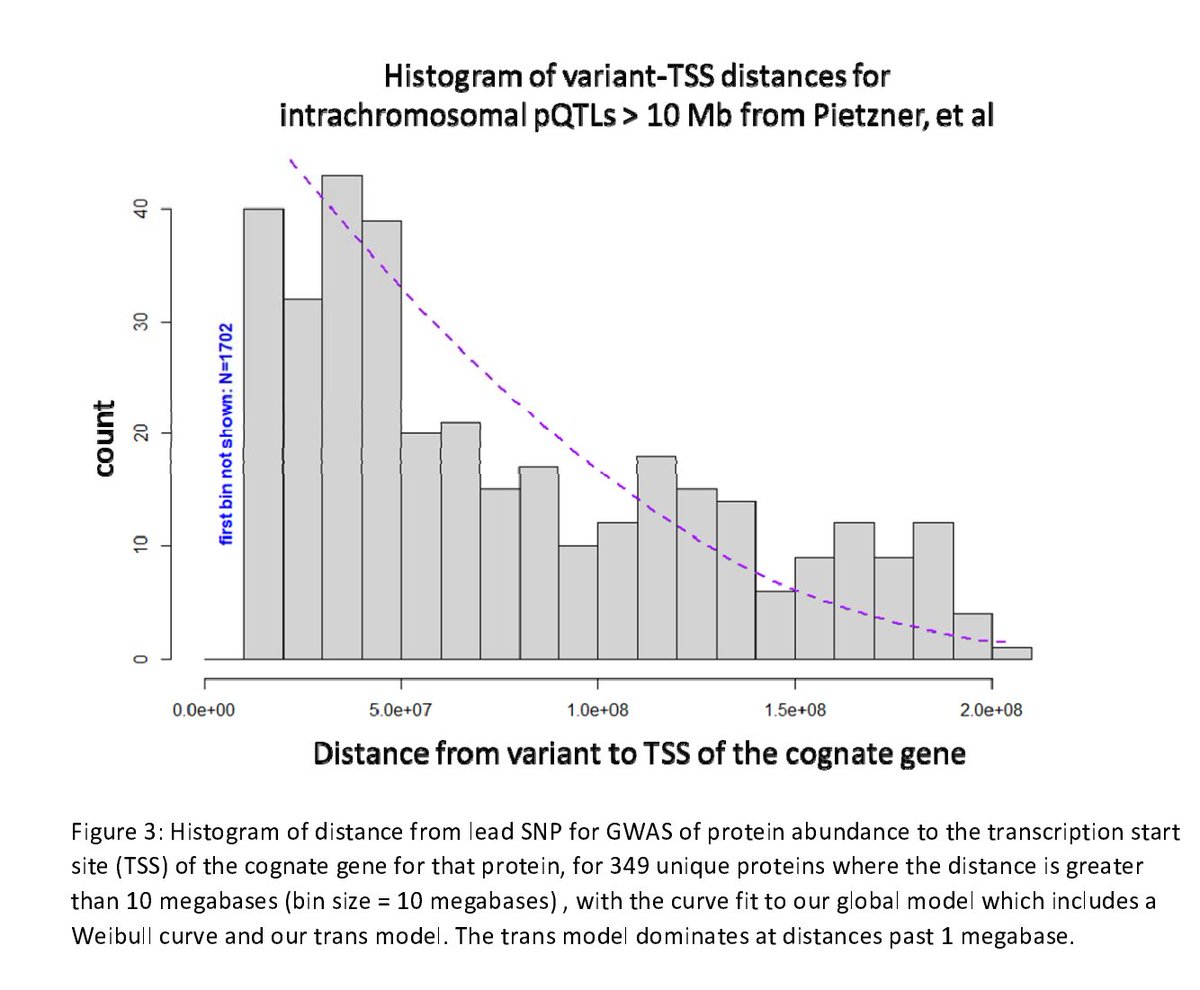


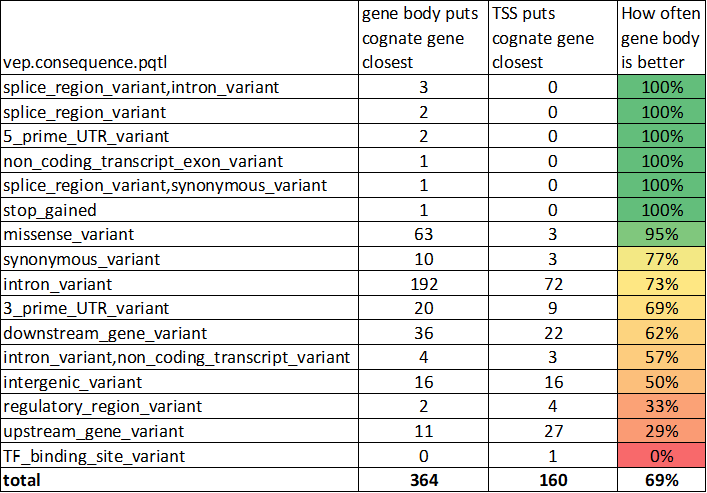
Email the whole thread instead of just a link!











