
Pfizer revealed great results for their COVID19 drug: prevention of 89% of hospitalizations if taken within 3 days of symptoms (85% if 5 days). I think you've all heard by now.
The drug blocks the coronavirus protease. As some of you know, my lab works on the same thing...
The drug blocks the coronavirus protease. As some of you know, my lab works on the same thing...
and in fact we have been testing the Pfizer drug (PF-07321332) in the lab, because it differs from our own previously revealed SARSCoV2 protease inhibitor (from September 2000) by only a few atoms, so we synthesized PF-07321332 as well.
https://twitter.com/michaelzlin/status/1379842117372026882?lang=en
Knowing Pfizer's compound (now called Paxlovid) works so well and was zipping through clinical trials (helps when you have $billions), we have been working on making even better protease inhibitors, hence our silence since September 2000.
But I can reveal a few things about how Paxlovid works and why it works so well. I can also tell you some info about how such drugs are designed, which it seems Pfizer will not reveal. But first, for reference, here's Pfizer's release
pfizer.com/news/press-rel…
pfizer.com/news/press-rel…
Now I'm booked thru Monday, I'll have to work on this thread later. In the meantime, the most important thing is: this does *not* mean vax are less necessary. Vax are way more effective as they suppress disease incidence, and we expect drug resistance will arise too. More later.
Typo above: we revealed our drug Sep 2020
Okay I’m back to explain the origins of Pfizer’s SARSCoV2 protease inhibitor, PF07321332 or Paxlovid. Sorry for the delay; had urgent grant deadlines to meet (grant proposals being a necessary but mostly wasteful academic activity, with current funding rates bouncing around 13%).
Coronavirus protease inhibitors are indeed a big deal in terms of treating disease in vulnerable people (but as with all antiviral drugs, really absolutely should *not* be considered an alternative to getting vaccinated).
The first question is if the protease inhibited by Pfizer’s drug is a good drug target. It's the best drug target we know of at the moment. As background, step 1 in SARSCoV2 replication is the translation of its RNA by host ribosomes to create the early viral polyprotein.
Step 2 is the cleavage of this polyprotein into smaller fragments by two viral proteases, a papain-like protease and a trypsin-like protease. Figure below from nature.com/articles/s4157… 

The trypsin-like protease is also called 3C-like protease (3CLpro) because it resembles the 3C protease of rhinoviruses that cause the common cold, or main protease (Mpro) because it is responsible for most processing events. In my lab we use the Mpro name.
Importantly, Mpro prefers to cleave after Gln residues, unlike any human protease. Below in (c) you can how a Gln side chain fits in a pocket of Mpro. This suggests the possibility of making drugs that preferentially inhibit Mpro over host proteases by having a part mimicking Gln

Even better, inhibitors of the related rhinovirus 3C protease had been developed that were orally bioavailable and capable of reducing disease in humans. This suggested that development of oral inhibitors for SARSCoV2 protease was within physical possibility.
Thus for the 3 reasons above —an absolute requirement for its activity in the viral lifecycle, unique specificity, precedent — Mpro is an excellent drug target.
How about the viral polymerase targeted by remdesivir and Merck/ridgeback’s molnupiravir? This can be either inhibited or made unreliable by nucleoside analogues. In the case of approved SARSCoV2 inhibitors remdesivir or molnupiravir, the MOA appears to be lethal mutagenesis:
i.e. the drug causes the polymerase to make so many mistakes that the viral progeny are deficient. While I have no proof my concerns can turn real, this mechanism of action seems to me to be concerning for an oral drug.
What will happen when patients are asked to take molnupiravir for a week? Some of these patients may think COVID19 is overblown, and maybe didn't seek treatment but were found to be infected during screening and then identified as at-risk.
Might it be possible that some people would take doses too late or skip doses? In such a case, viruses will spend some time in a low concentration of drug, too low to block viral replication but not zero either. In such a case you can expect a non-lethal rate of mutation...
But that rate will be far higher than the natural rate. And then in those patients with poor compliance who indeed don't fight off the virus, then the virus will still replicate to high levels and spread to others...with mutations.
This is again just a theoretical concern, but I haven't seen it raised yet. It seems to me to be a recipe for supercharging viral replication. I don’t have this problem with remdesivir, because that’s given by IV drip in the hospital. You can be sure it will be dosed correctly.
Thus lethal mutagenesis just does not seem like a safe mechanism of action for oral drugs given to a some people who are doubtful of the severity of the diagnosis to begin with. This is distinct from the other mutagenesis-related concern over molnupiravir...
which is if molnupravir might also be mutagenic to human DNA. That would obviously be a no-go, but Merck reported no mutagenesis in mammals in vivo, which apparently satisfied the UK’s MHRA. The debate is summed up here: drugdiscoverytrends.com/early-safety-c…
But perhaps most irrefutable is that molnupiravir only prevents hospitalization by 50%, when there are other drugs that work far better without any risk whatsoever of viral or human mutation, such as the monoclonal antibodies from Regeneron or Vir, and now PF07321332.
Actually since molnupiravir only stops hospitalization 50% of the time, what is happening to the virus the other 50% of the time. Has Merck sequenced those escaped viruses to see if they have higher rates of mutations?
Anyway that's it for today. Will continue another time.
Continuing the protease inhibitor thread. Above we discussed why the SARSCoV2 protease is a good target for the development of oral antiviral drugs. So why would we want to have an oral antiviral drug? It's worth presenting the rationale to make sure efforts are well allocated.
The idea is to have an antiviral that could be given to vulnerable patients, e.g. elderly or immunocompromised, to prevent breakthrough infections from becoming breakthrough hospitalizations. This already exists with monoclonal antibodies, but these are perishable protein drugs.
mAbs are expensive to make and need refrigeration, and thus harder to stock. You then have to find those clinics, which may be far away, and you have to get them injected IV. Nobody likes getting an IV line put in, so many will avoid the hassle and pain.
Oral pills can be delivered overnight in regular packaging wherever they are needed, and they are painless to take.
Given the hype over Pfizer and Merck, it seems people don't need convincing about oral drugs, but it's nice to make clear what those merits actually are.
Given the hype over Pfizer and Merck, it seems people don't need convincing about oral drugs, but it's nice to make clear what those merits actually are.
Before SARSCoV2 appeared, my lab was already working with trypsin-family viral proteases from another positive-strand RNA virus, hepatitis C virus (HCV). We were also working with its clinically approved orally available inhibitors, including one called boceprevir (BPV).
As a postdoc with Roger Tsien at UCSD, I introduced the use of HCV protease inhibitors to control protein structure and function in cells via turning off HCV protease. Thus I had developed familiarity with the atomic details of drug-protease interactions
pnas.org/content/105/22…
pnas.org/content/105/22…
We expanded this to allow drug control of other previously undruggable RNA viruses used for vaccines and gene therapy. We thus borrowed, for gene therapy purposes, orally available drugs developed for $billions by pharma companies, for essentially free.
nature.com/articles/nchem…
nature.com/articles/nchem…
Most recently we mutated HCV protease to make two variants that were susceptible to different sets of HCV protease inhibitors, which gave us even more knowledge about the mechanisms of drug binding.
nature.com/articles/s4159…
nature.com/articles/s4159…
That's why when SARSCoV2 appeared, and I looked at the atomic structure of SARSCoV1 Mpro (>90% identical to SARSCoV2 Mpro), I happened to notice that it was very similar in structure to HCV protease. I was even more shocked when I took BPV, a FDA-approved oral drug, and..
using a protein-viewing program, I docked BPV virtually into the active site of SARSCoV1 Mpro (and later SARSCoV2 Mpro, when its very similar structure was published). To my surprise, it was a nearly perfect fit. Here, BPV is blue, Mpro is gray, and a natural substrate is yellow.
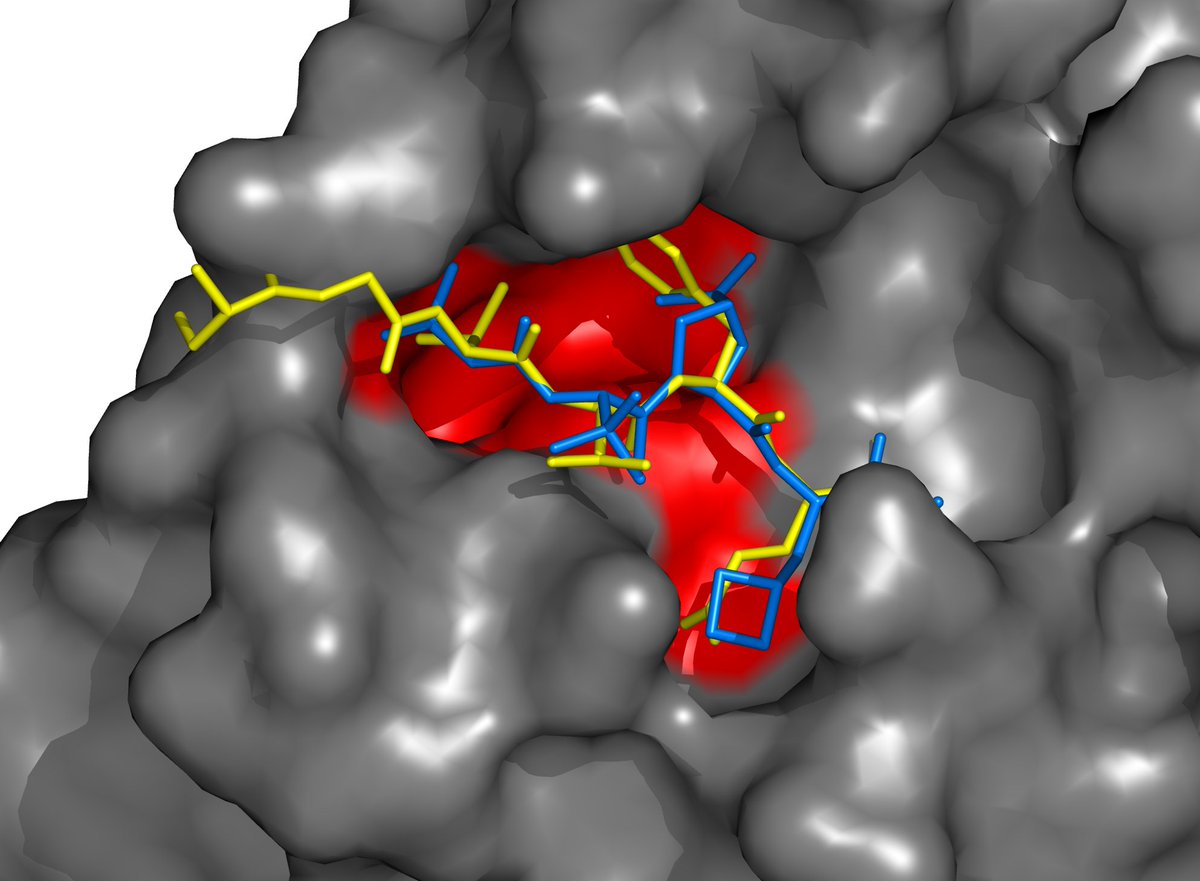
It was amazing... here we have an already FDA-approved oral drug fitting SARSCoV Mpro pretty well. Weirdly, there were already reports of tests of existing antivirals (including many with HIV protease inhibitors), both in reality and virtually, and none mentioned BPV.
So we just had to test it ourselves. We synthesized the gene for SARSCoV2 Mpro, engineered a membrane-localized GFP tethered to the plasma membrane via a SARSCoV2 Mpro substrate, and expressed both in mammalian cells. Amazingly, we saw that BPV at just 1μM inhibited Mpro function

We realized BPV itself would not be good enough, as we want inhibition at <1μM, and BPV wasn't so effective in lung cells. But we had the idea of modifying BPV to improve its fit to SARSCoV2 Mpro.
One idea we had was to incorporate a Gln-mimicking group used previously in rhinovirus 3CL inhibitors, to tighten the binding to Mpro. You recall above how Mpro prefers Gln at the last position before the cut site. HCV prefers a different set, and so BPV didn't mimic Gln there.
With this idea, we applied for a tiny bit of funding from BSAC, a UC COVID emergency initiative, FastGrants from @patrickc, and Stanford Chem-H (<$50k each). FastGrants and Stanford Chem-H came through, thank you!
Soon after our finding, other groups also announced that BPV could inhibit SARSCoV2 Mpro modestly well, verifying our results. By then we had already started synthesis on our designs for improved drugs.
We had a novel theory for why BPV worked so well. As mentioned above, Mpro prefers Gln at the 1st position before the cut site (P1), but HCV protease doesn't, so BPV's P1-analogous group is not a Gln analog.
Conversely, BPV has a Pro analog at P2 to match HCV protease's preference, but Mpro only takes substrates with Leu or Phe there. We hypothesized BPV's Pro analog fits Mpro because it contains extra atoms to mimic Leu, while constraining BPV's backbone into the right conformation.
So we wanted to preserve BPV's Pro analog at P2, while optimizing P1 for Mpro. We synthesized a drug called ML1000 that incorporates a Gln mimic in the first position before the cut site, but otherwise is similar to BPV. This was the first described combination of these elements.
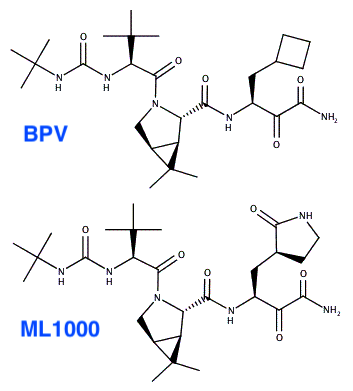
We found this drug (labelled compound 1 below) to improve inhibition of Mpro in enzymatic and cellular assays. An additional modification 2 positions before the cleavage site (compound 2) worked similarly. We posted our findings on biorxiv in 9/2020. biorxiv.org/content/10.110…

Pfizer revealed the structure of PF-07321332 in April 2021. As you can see below, it incorporates the same two features that we identified as most important: The P2 Pro analog of BPV and the P1 Gln analog. It also has the P3 t-butyl group of BPV.
https://twitter.com/michaelzlin/status/1379842117372026882
What's interesting is that when Pfizer finally published their manuscript describing PF-07321332, it did not say the P2 and P3 groups were copied directly from BPV. In fact it didn't mention BPV at all! science.org/doi/10.1126/sc…
Now I don't know why Pfizer wouldn't mention BPV. Maybe it wanted to pretend its designs were 100% original, or maybe it didn't want to invite a charge of patent infringement from Merck, who acquired ScheringPlough which developed BPV, even though BPV is off-patent. Anyway...
What's most notably different in PF-07321332 from BPV and ML1001 is the group that reacts with the active site cysteine of Mpro. This group is known as the warhead. Pfizer uses a nitrile (CN) while BPV and ML1000 use a ketoamide (COCONH2).

Pfizer's use of a nitrile made PF-07321332 more cell-permeable and more orally bioavailable than ML1000. We know because we have synthesized PF-07321332 and tested it alongside our drugs.
Pfizer didn't say how many molecules they tested to find the best combination of elements, but it's certainly more than the handful they reported. It's probably hundreds, and probably dozens tested for bioavailability in animals.
In contrast, we could only afford synthesizing <20 and testing 4 drugs in mice so far, and that's after another generous infusion of funds from FastGrants and Stanford and the Coulter Foundation (~$100k each).
Since we knew Pfizer was already in clinical trials with PF-07321332 (which they carried out with impressive speed — something they have done well throughout the epidemic) we have been concentrating on making the next generation of drugs.
PF-07321332 is not a perfect drug. It makes sacrifices for its oral permeability. It's degraded easily, requiring coadministration of ritonavir to suppress drug-degrading liver enzymes, thus causing drug-drug interactions. Also, the PF-07321332 dissociates from Mpro quite quickly
Now that PF-07321332 is available, we are relieved that a good option will finally be available for vulnerable vaccinated patients who get sick. But if the pandemic has taught us anything, it's to never count out SARSCoV2. Thus we are continuing to work on our inhibitors.
We recently obtained promising results with a compound with nearly equivalent efficacy as PF-07321332 against virus that is also more stable, and we have also explored alternative structures as a hedge against viruses becoming resistant to PF-07321332.
Just as with HIV and HCV, what's likely to happen is that multiple labs and companies will develop multiple effective Mpro inhibitors. This will provide a more diverse arsenal to counteract resistance, and the competition will lower drug prices.
Severe disease from HIV and HCV is now rare, in part because of protease inhibitors (combined with nucleoside analogs). I am optimistic that severe disease from SARSCoV2 can be a rare thing in the future as well.
🙏acknowledgements for what continues to be an incredible team effort:
• funders above
• lab members designing our newest drugs and doing the very hard work of rigorous testing: Michael Westberg, Yichi Su, and Xinzhi Zou
• collaborator and real COVID19 expert Catherine Blish
• funders above
• lab members designing our newest drugs and doing the very hard work of rigorous testing: Michael Westberg, Yichi Su, and Xinzhi Zou
• collaborator and real COVID19 expert Catherine Blish
We expect to report our latest drug designs soon, and then we'll need to seek more funding. This time, I'm hoping NIH reviewers will be willing to provide just a little bit of the $3B of funding earmarked for academic research on antiviral meds:
nytimes.com/2021/06/17/hea…
nytimes.com/2021/06/17/hea…
P.S. have been asked a few times why you can't skip vaccines and take drugs. Many many reasons. If you didn't get vaxxed, you *will* get COVID19 sooner or later (sooner if many others don't get vaxxed too). This infection would then be super-problematic for you and others.
1. For you, you would need to be tested right away upon getting symptoms to get the drug. You might not think you have COVID19 or you might feel yourself getting better, only to land up in the hospital and be too late for treatment.
2. For you, you might not be treated anyway if you're low risk, but then you might be unlucky and land up in the hospital anyway or with long COVID.
3. For you, you might be traveling/vacationing/camping and not have easy access to testing when you get sick, or not have access to the drug where you are.
4. For others, the disease is most contagious the day *before* you get symptoms, or you might never develop any symptoms at all and still be contagious at some time. So then you can give the disease to someone vulnerable, when then may not get diagnosed in time to get treated.
5. If everyone acted as irresponsibly as you by turning down vaccines to wait for treatment, then in addition to all the above occurring a million times, the virus gets millions more chances to mutate higher contagiousness and *drug resistance* thereby setting us back to square 1
So it's clear antivirals are only useful when used in limited targeted ways. They are in no way whatsover a substitute for vaccines, not even for 1 single person.
I probably missed several other good reasons, but the above are sufficient reasons. I hope this explanation is clear
I probably missed several other good reasons, but the above are sufficient reasons. I hope this explanation is clear
And finally I will point out the irony of taking a pro-drug anti-vaccine stance.
Vaccines are the most natural way to prevent infection: you expose your immune system to a viral protein to teach it, so that when you get the virus, you mount much faster the same type of response you would have mounted if you weren't vaccinated.
Drugs on the other hand are human inventions that did not exist in nature previously, and very few drugs are perfect. Most drugs have some side effect or another, because they will interact with proteins in the body and not just with viral proteins.
Before the pandemic, pharma companies were criticized for not spending enough funding on vaccines. They were accused of preferring expensive drugs ($100s to $100,000s per case) over preventing disease with inexpensive vaccines ($50 per patient).
Now we have good vaccines, and some people don't want to take the 2-time vaccination, but would rather get sick, get tested, put their close contacts in quarantine, isolate and miss work and social gatherings, get others sick, and take drugs for a week...
Well that's interesting. Pardes has a patent that apparently covers Pfizer's Paxlovid (PF-07321332). Notice the granted invention encompassing Paxlovid. We may see a big licensing deal or courtroom drama in the future.
Interestingly, the patent doesn't cover our compounds
Interestingly, the patent doesn't cover our compounds

For the non-chemists and non-biochemists: Marking the H's is optional of course, and R means anything
• • •
Missing some Tweet in this thread? You can try to
force a refresh









