
What happens to BNP when a person is started on #sacubitrilvalsartan?
#BNP should go up, right?
Thanks to work by @pmyhre in our group, we have some new and interesting data.
A 🧵, read on! 👇
#BNP should go up, right?
Thanks to work by @pmyhre in our group, we have some new and interesting data.
A 🧵, read on! 👇
When the #PARADIGM-HF study was published, one of the things immediately noticed was the increase in BNP that occurred after being started on sacubitril/valsartan.
This is because through effect of sacubitril, neprilysin is inhibited. Why might this lead to an increase in BNP?
This is because through effect of sacubitril, neprilysin is inhibited. Why might this lead to an increase in BNP?

#Neprilysin is a ubiquitous metalloproteinase that assaults BNP (among other targets including ANP and CNP) and cleaves it in numerous places as shown.
Note that in BNP, there are cleavage sites that involve numerous places where immunoassays for measurement of BNP may bind.
Note that in BNP, there are cleavage sites that involve numerous places where immunoassays for measurement of BNP may bind.
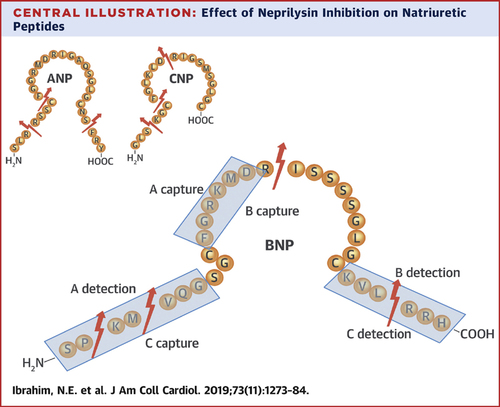
Thus, the results from PARADIGM seemed to make sense: as published by @pmyhre, BNP on average by 19%, doubling in 18%, and tripling in 6%.
#NTproBNP is not a target for nep, making it a superb monitoring tools for prognosis when using sacubitril/valsartan.
#NTproBNP is not a target for nep, making it a superb monitoring tools for prognosis when using sacubitril/valsartan.
Therefore, most that measure biomarkers in heart failure agree that NT-proBNP is the preferred means by which to monitor prognosis and longitudinal remodeling status in those treated with sacubitril/valsartan.
Policy documents and guidelines say this as well.
But there's more!
Policy documents and guidelines say this as well.
But there's more!
After PARADIGM-HF, word began to circulate that with some frequency some patients treated with sacubitril/valsartan did not show the "expected" rise in BNP.
We decided to examine this question. To do so, we utilized the resources of the #PROVEHF and #EVALUATEHF studies.
We decided to examine this question. To do so, we utilized the resources of the #PROVEHF and #EVALUATEHF studies.

In this analysis, we examined BNP changes from initiation/titration of sacubitril/valsartan, using the Siemens BNP method (the same one from PARADIGM).
And we found: ZERO increase in BNP. NT-proBNP fell by 30%.
And we found: ZERO increase in BNP. NT-proBNP fell by 30%.

Across all the concentration changes of BNP and NT-proBNP, the difference was the same: 30%. So if NT-proBNP went down by 50%, BNP fell by 20%. If NT-proBNP went up by 10%, BNP rose by 10%.

Predictors of BNP increase were predictable: worse clinical picture, lower BMI, AF, etc.
Stated another way, increase in BNP in this analysis was pegged to prognosis.
Stated another way, increase in BNP in this analysis was pegged to prognosis.

"But wait a minute" you might be saying, "you told me that neprilysin degrades BNP, so why isn't it rising?"
The 30% offset from NT-proBNP tells the story: indeed, BNP is RISING due to nep inhibition...however due to down regulation of the BNP gene, this offsets the rise.
The 30% offset from NT-proBNP tells the story: indeed, BNP is RISING due to nep inhibition...however due to down regulation of the BNP gene, this offsets the rise.
How do we reconcile different results from PARADIGM?
In both PROVE and EVALUATE, we did not have an NT-proBNP inclusion criterion, did not have a run in period, and did not remove individuals that could not reach target doses. This may have something to do with it.
In both PROVE and EVALUATE, we did not have an NT-proBNP inclusion criterion, did not have a run in period, and did not remove individuals that could not reach target doses. This may have something to do with it.
What do these results mean?
First, one should take away from this is the fact that lack of rise in BNP after S/V treatment should NOT be inferred as a "lack of effect" from neprilysin inhibition.
It also means that BNP rise is unlikely to explain all the benefit of S/V.
First, one should take away from this is the fact that lack of rise in BNP after S/V treatment should NOT be inferred as a "lack of effect" from neprilysin inhibition.
It also means that BNP rise is unlikely to explain all the benefit of S/V.
What neprilysin target might explain the suppression of the BNP gene and rise in cyclic GMP?
We previously reported a HUGE rise in #ANP from these same samples. ANP is a major target for neprilysin and drove reverse remodeling in PROVE-HF.
jacc.org/doi/10.1016/j.…
We previously reported a HUGE rise in #ANP from these same samples. ANP is a major target for neprilysin and drove reverse remodeling in PROVE-HF.
jacc.org/doi/10.1016/j.…

So, can BNP be used to monitor persons treated with sacubitril/valsartan?
Yes. Yes it can. But it may be tricky. So these results do not change the current recommendations that NT-proBNP is preferred for this application whenever possible.
One last question...
Yes. Yes it can. But it may be tricky. So these results do not change the current recommendations that NT-proBNP is preferred for this application whenever possible.
One last question...
In PARADIGM and this analysis, the Siemens BNP assay was used. Since neprilysin cleaves BNP in multiple places where different immunoassays may bind, is it possible that sacubitril/valsartan differentially affects BNP if measured by assays using different antibody binding sites?

Stay tuned...more #PROVEHF data are coming soon!
I want to thank @pmyhre for his hard work and look forward to continuing to clarify the story.
And remember everyone: #GDMTworks
I want to thank @pmyhre for his hard work and look forward to continuing to clarify the story.
And remember everyone: #GDMTworks
• • •
Missing some Tweet in this thread? You can try to
force a refresh


