
Finally published in @naturemethods: CellRank for directed #SingleCell fate mapping, our take on studying cellular dynamics using RNA velocity. Fantastic collab between labs of @dana_peer @sloan_kettering and @fabian_theis @HelmholtzMunich, read at go.nature.com/3Fm17lt. 1/17
Motivation: scRNA-seq is great to study cellular heterogeneity, but its application to continuous processes is limited by the fact that cells are destroyed upon sequencing. Hence, we obtain many genes across many cells, but only through static snapshots. 2/17
Trajectory inference algorithms have been developed to reconstruct dynamics from static snapshots. They order cells according to similarity and even detect branches. But what about the direction? Often unclear, esp. in perturbed settings like regeneration, reprogramming etc. 3/17 

A powerful solution is RNA velocity @GioeleLaManno which uses the ratio of spliced/unspliced counts to infer gene up/down reg. Led by @VolkerBergen, we generalized the concept with scVelo.org, however, how do to interpret the high-dim., noisy vector fields? 4/17

We developed CellRank.org for this task, which reconstructs dynamics based on RNA velocity and exp. similarity in high dimensions. Our aims: robustness to noise, stochastic formulation, highly scalable and able to capture gradual nature of fate commitment. 5/17

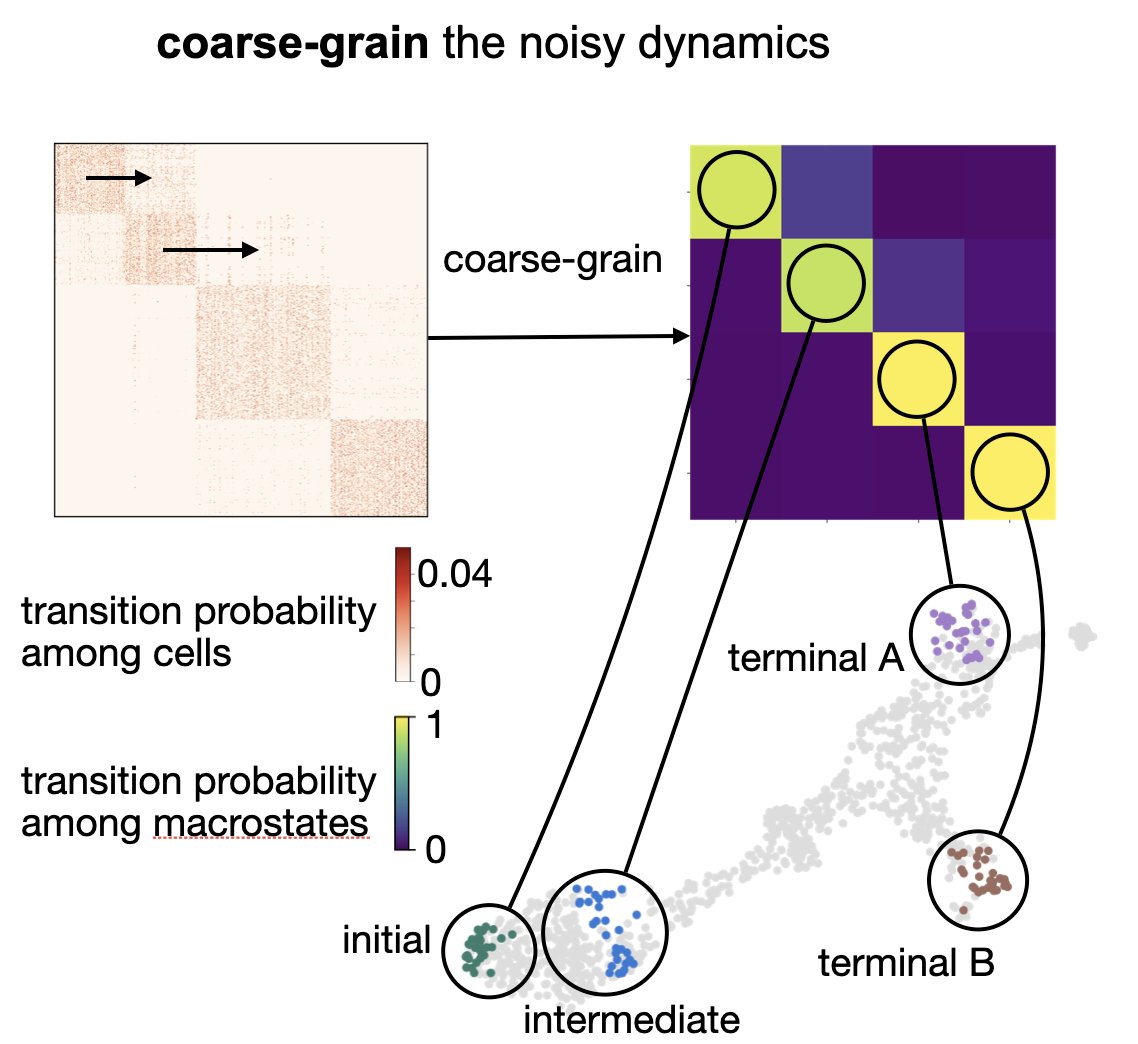
Algorithm: set up a Markov chain to describe cell-state changes. KNN graph construction so that transitions are restricted to nearest neighbors and compute correlation of velocity vectors with nearest neighbors -> T. Propagate velocity uncert. into transitions (not shown). 6/17

We teamed up with Bernhard Reuter to coarse-grain T by adapting GPCCA to the single-cell context. This gives us macrostates and transitions among them -> identify initial, intermediate and terminal states. Available in CellRank through pyGPCCA: bit.ly/pygpcca. 7/17
How likely is each cell to reach each terminal state? CellRank finds out by computing smooth fate probabilities. To infer putative decision driver genes, we correlate these against gene expression. 8/17

Using fate probabilities, CellRank charts smooth, trajectory specific gene expression trends in any precomputed pseudotime. Of course, you can use CellRank's detected initial state to root that pseudotime. 9/17

Application: we started with validation on @morris_lab in-vitro reprogramming data which has CellTagging lineage barcodes. Using these as ground truth, we show that CellRank successfully predicts reprogramming outcome. 10/17

Moving beyond in-vitro, we applied CellRank to pancreas dev. at E15.5 where we correctly predict initial & terminal states, comp. fate probs and recover exp. trends of the main drivers. Collab @bakhti_mostafa from @LickertHeiko lab. 11/17

To demonstrate robustness, we varied key method parameters and re-computed fate probabilities. For each parameter and trajectory, we correlated fate probabilities for different parameter value pairs. 12/17

Benchmarking: How do competing methods perform on the pancreas data, with and without RNA velocity information? CellRank is the only method to correctly predict all of initial & terminal states, fate probs and driver gene exp. trends. 13/17

Scalability is key to us - thanks to Michal Klein's implementation magic, CellRank runs on 100k cells in <2 min while being extremely memory efficient. 14/17

Finally, we demonstrate that CellRank generalizes beyond normal development on lung regeneration data where it predicted a novel dedifferentiation trajectory, collab @schniering_j and @Meshal_Ansari from @SchillerLab. 15/17

If this dedifferentiation trajectory exists, we would expect to find novel intermediate cell states between Goblet and Basal cells after injury but not in control mice; that's exactly what @schniering_j found in immunofluorescence stainings. 16/17

Wrap up: CellRank.org is a scalable, easy to use framework to compute directed cell-state trajectories. Massive thanks to co-authors I couldn't mention above, @VolkerBergen & @SettyM, and thanks for support from @BayerFoundation, @jherzstiftung, @MunichQbm. 17/17
• • •
Missing some Tweet in this thread? You can try to
force a refresh





