
Delighted to introduce FEgrow: An Open-Source Molecular Builder and Free Energy Preparation Workflow! By @bieniekmat @ChemCree @rachaelpirie203 @joshhorton93 @drnataliej
Now out on @ChemRxiv #compchem: doi.org/10.26434/chemr…
🧵tweetorial...
Now out on @ChemRxiv #compchem: doi.org/10.26434/chemr…
🧵tweetorial...
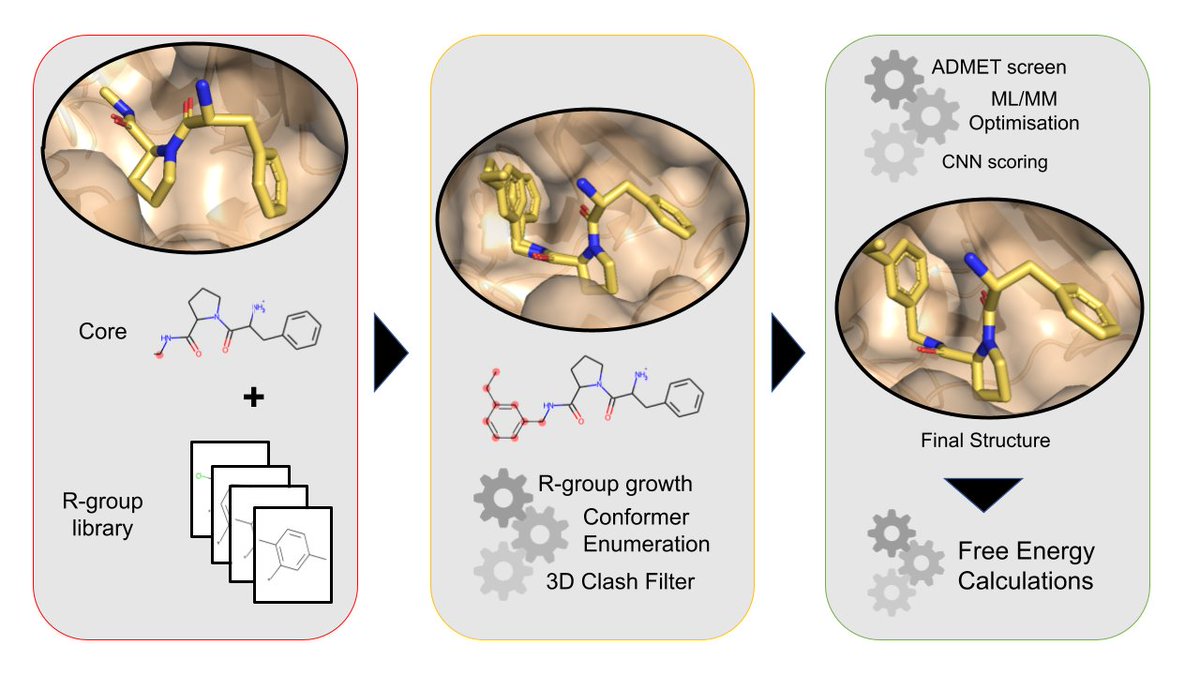
Let's say we have the structure of a hit compound and want to build a congeneric series of ligands bound to a protein. A common task in free energy setup.
For our example, we'll take structures from this very nice study by @JorgensenWL & coworkers: doi.org/10.1021/acscen…
For our example, we'll take structures from this very nice study by @JorgensenWL & coworkers: doi.org/10.1021/acscen…
We can use the very cool mols2grid package to interactively select functional groups to add to the core from a library.

We can use @RDKit_org to attach the functional groups, enumerate the conformers and filter out the ones that clash with the protein.

Which remaining conformer is lowest in energy? We perform structural optimisation using a hybrid machine learning / MM potential (with the ANI2x deep learning potential) in @openmm_toolkit.

And optionally output simple ADMET properties and binding affinity predictions for the final structures using the very user-friendly gnina convolutional neural network scoring function: github.com/gnina/gnina
We've built several of the Mpro inhibitors using this workflow, and computed relative binding free energies with biosimspace/SOMD (biosimspace.org), with pretty good agreement with experiment.

Many thanks to all the open science software, blogs and tutorials that went into this workflow. They're hopefully listed in full here: cole-group.github.io/FEgrow/acknowl…
This was a bit of a fun, side-project for us, but we hope it's useful to others too. Feel free to send comments, issues & feature requests.
code+tutorial: github.com/cole-group/FEg…
docs: cole-group.github.io/FEgrow/
code+tutorial: github.com/cole-group/FEg…
docs: cole-group.github.io/FEgrow/
• • •
Missing some Tweet in this thread? You can try to
force a refresh


