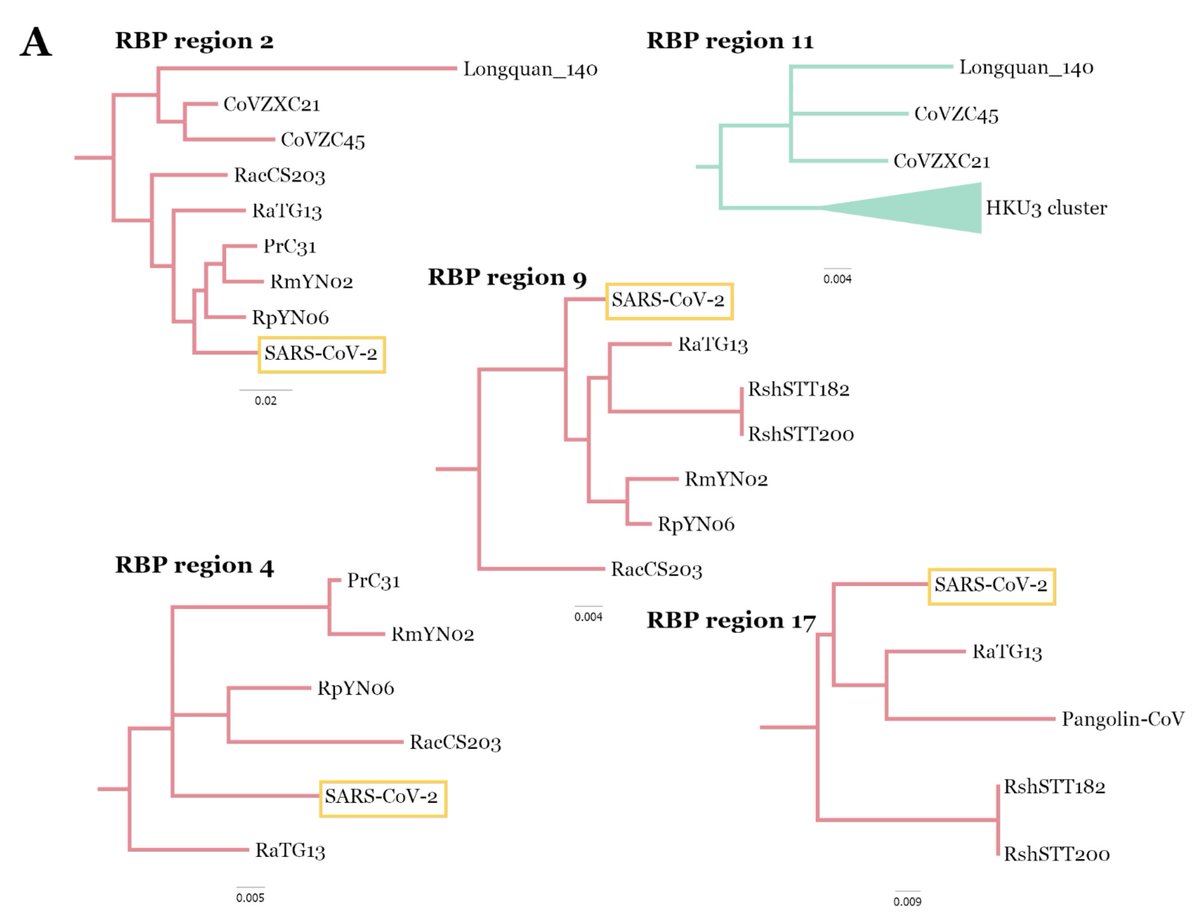
Recently I've been too busy to engage with the latest cuckoo discussions on SARS-CoV-2 origins, but I wanted to comment on the Senator Burr report that's making the rounds, especially since they use a figure I made (!) for their fig2 help.senate.gov/imo/media/doc/…
First things first, I was never consulted about this report or had any knowledge of it before the last 24 hours.
To my understanding this is a US senate commissioned report assessing the origins of SC2. Not sure who actually wrote it (don't really care), but they've done a very VERY bad job at reporting the science on this topic... here's some thoughts:
1. The report seems to assume that my figure 2 phylogeny is the single way these viruses are related, but this isn't the case. this tree is derived from only one non-recombinant bit of the viruses' genomes. 

In fact, the GBE paper shown at the bottom of fig2 (but not referenced or mentioned in the report's text) goes into great depth about how important recombination is and how every chunk of the viruses genomes has a very different evolutionary history: doi.org/10.1093/gbe/ev…
2. The report states how 'the earliest variants of SARS-CoV-2 were well-adapted for human-to-human transmission' and how this 'represent a significant break from the precedent of other zoonotic spillovers involving respiratory viruses', but this is largely misleading...
one example of many other large-scale zoonotic human pandemics being very 'well-adapted for human-to-human transmission' is the avian flu 1918 pandemic and swine flu 2009 pandemic...
I mention this since we recently discovered that a single amino acid change at flu's NP protein can evade an important human-specific immune response and that change has happened in both 1918 and 2009 independently: doi.org/10.1101/2022.0…
These 'human-adapted' changes however most likely happened in the respective animal hosts BEFORE the viruses spilled over into humans (with the earliest known 1918 pandemic sequences having the human-adapted residue: nature.com/articles/s4146…)
this is only one of many examples where changes that increase infectivity in a host take place before the host switch (if 'human-adapted' changes had to be engineered in a lab we wouldn't have any zoonotic pandemics anyway...)
So, the report is being rather disingenuous about how different the COVID-19 pandemic is to other historical large-scale pandemics.
3. There is a rather odd comparison between the COVID-19 pandemic and the H7N9 flu epidemics in China
The H7N9 viruses circulate in birds many of which are farmed and come into very frequent and close contact with humans and in China there's been at least 5 H7N9 epidemics from independent sources documented in the last 10 years doi.org/10.1016/S1473-…
this is a different story to SARS-CoV-2's essentially one-off spillover. The genetic data support that SC2 was introduced in humans in 2 spillover events, BUT these 2 events happened at almost the same time from a single host population source: doi.org/10.1126/scienc…
thus the comparison to different H7N9 viruses spilling over from farmed birds repeatedly but months apart is irrelevant (not to mention the date typos that make you wonder if anyone proof-read this report...)

4. My final point is about how similar our understanding of the SARS-CoV-1 emergence origins is compared to that of SC2, since the report makes it sound like we know everything about SC1 and nothing about SC2 origins...
We recently performed a very comprehensive analysis to see, once you account for the complex recombination patterns in these viruses, how close sampled animal sarbecoviruses are to SC1 and SC2 respectively: virological.org/t/the-comparat…
what we find is that the recombination-free closest common ancestor (recCA) to each virus is basically of equal identity ~99%, while the whole-genome similarity between SC2 and its closest bat virus (~97%) is higher than that of SC1 and its closest bat virus (~96%)

All in all, this report cherry picks data and in many cases makes assertions that completely contradict the scientific facts and data that are available.
It also provides no actual evidence for a lab origin of the virus (WIV just being in Wuhan is NOT real evidence of a lab leak) and contains plenty of political 'US vs China' discussion points (that imho obstruct the scientific search for the pandemic's origins).
• • •
Missing some Tweet in this thread? You can try to
force a refresh