
Doing #SingleCell #RNAseq? Ever wonder if all those clusters are real? Turns out most feature selection & clustering pipelines can't tell when there's only 1 cluster! But I found a solution! 🧵👇 

Happy to release (and welcome feedback!) on my new feature selection algorithm that can help prevent false discoveries in scRNAseq datasets! bitbucket.org/scottyler892/a… (pip installable & works easily with scanpy :-) @fabian_theis
@fabian_theis By starting from first principles I asked: what is a cell type? We've long identified them as subsets of cells with different functions that have corresponding marker gene expression

@fabian_theis If this is true, we can find them algorithmically by looking specifically for anti-correlation patterns! Indeed, looking at a dummy pancreatic dataset, this was true for both specific cell types and also broad lineage markers.

@fabian_theis And if you only have one meaningful subset present, that marker gene's negative correlation pattern disappears! So - in theory a gene should only have an excess of negative correlations if there are multiple cell identities present. (Also in practice!)


@fabian_theis To test if a gene has "too many" negative correlations, I came up with an FDR inspired algorithm, measuring the observed number of negative correlations compared to what's expected from a shuffled dataset.
@fabian_theis So: does it actually pass the "null dataset" test? It does 😏, where other approaches don't, and regardless of clustering algorithm. (0 features returned when there are no meaningful groups!)

@fabian_theis In the single cell field I've met some people who have told me that they don't think negative controls are important for our field. But this of course has uncomfortable downstream consequences. But why is this a real world problem too?
@fabian_theis Sub-clustering... I've seen papers doing recursive sub clustering - which is totally fine! But it would be really great if we had an algorithm that could prevent a false discovery before a bench biologist has to spend years 🗓️ and 💰 to do it (I've seen this happen 😞).
@fabian_theis When sub-clustering, if you keep on dividing a dataset, you'll hit a point where you're subdividing based on Poisson noise. In cell-lines it'll probably be after 1 round. How many clusters do you get with each method though? (Wayyy too many! Unless you use anti-cor. FS approach)


So if your pipeline lets you divide ad infinitum - we can be sure that at some point, it changed to false discoveries. We can this the "recursion to completion" problem. Anti-correlation based feature selection passes this test too! (In both UMI & full-length based techs)



But having low false discovery isn't the only concern. We want to also be sensitive and find an the real populations! But for that we need a "ground truth" so have to use simulation. We want to find identify DEGs as our features! So how do the algorithms do?
Anti-correlation FS was at the top for 6/8 metrics! (But sensitivity was average :-/). But why? Turns out splatter's simulations make every gene highly expressed in every cluster (but still with DE across clusters) 🤔, blunting the anti-correlation pattern.


What about a second gen simulator like SERGIO that's based on a gene regulatory network structure? sciencedirect.com/science/articl… Using their simulator, our algorithm was the best performer in every 👏single👏 category 💥! Including sensitivity!! 🤯

One problem with doing all these correlations though is time... How well does it scale? My implementation works completely out of memory and can analyze the 1/4million cell tabula muris senis in 60 min (the clustering took 4 days!)

And using subclustering we were able to re-identify the PP cells of the pancreas (which were only 0.01% of the original dataset!!)
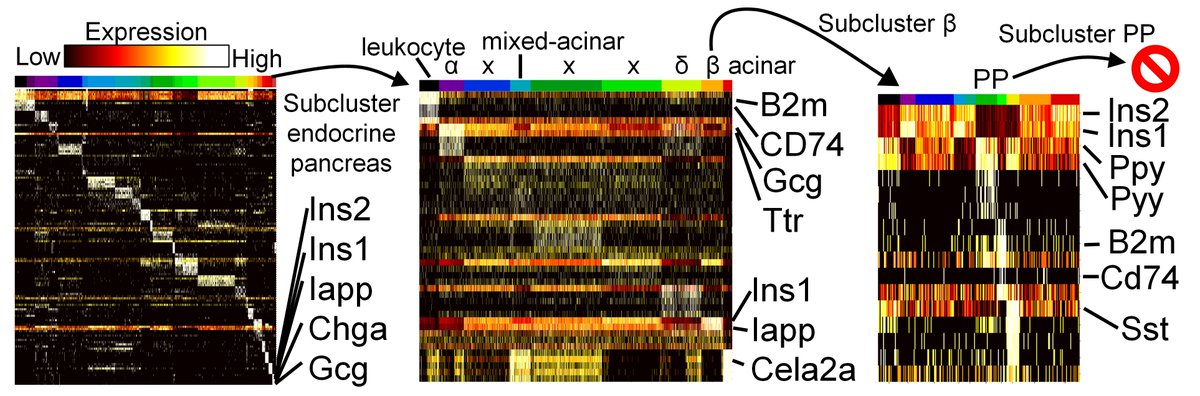
What's the take home? Computational negative controls can save us bench biologists a lot of 🗓️&💰, and push us to figure out new algorithms that can pass negative control tests. Anti-correlation based feature selection can answer the question "should I sub-cluster these cells?"
It also improves on feature selection efficacy over the standard HVG approaches! It's simple software to use, just give it the matrix, the gene names, and the species, and you get a nice pandas data frame of annotations!
I hope you all find my work valuable! It's a problem that I sat with for a really long time, and I'm quite happy with this solution =). Give it a try and see how it does on your data! (Brief tutorials on the host site!) =) pip install anticor_features
bitbucket.org/scottyler892/a…
bitbucket.org/scottyler892/a…
And because I forgot to link it at the top 🤦♂️ - Check out the pre-print here: biorxiv.org/content/10.110…
• • •
Missing some Tweet in this thread? You can try to
force a refresh



