1/16 I recently gave a public lecture about SARS-CoV-2 origins that touched on some of the myths & misinformation promoted by some proponents of a lab leak origin (below).
But I'd like to discuss a couple others here that I didn't have time to get into.
But I'd like to discuss a couple others here that I didn't have time to get into.
2/16 First, there is the talking point you've probably heard many times that there is something inexplicable about a new SARS-like coronavirus emerging in Wuhan because it is *so* far away from where the ultimate progenitor in bats existed.
3/16 But recombinant viruses related to SARS2 have been found in Hubei province, where Wuhan is.
And, leaving that aside, the fragment of the SARS2 genome most closely related to a known bat virus shares an ancestor with it only a few years before 2019...
And, leaving that aside, the fragment of the SARS2 genome most closely related to a known bat virus shares an ancestor with it only a few years before 2019...

4/16 (See "non-recombinant region" 3 in the above figure, from the virological.org post below from my colleagues and me.)
And it turns out the most closely related fragment is from a bat virus sampled in Yunnan province in southern China...
virological.org/t/the-comparat…
And it turns out the most closely related fragment is from a bat virus sampled in Yunnan province in southern China...
virological.org/t/the-comparat…
5/16 Not Laos, by the way.
So those who had shifted their goalposts for a lab origin to WIV-imported viruses from Laos will now need to shift them back to southern China I guess.
At least until the next scientific finding necessitates digging the goalposts up yet again.
So those who had shifted their goalposts for a lab origin to WIV-imported viruses from Laos will now need to shift them back to southern China I guess.
At least until the next scientific finding necessitates digging the goalposts up yet again.
6/16 Anyway, the key point: the distance travelled, potentially via the wildlife trade, between a possible Yunnan reservoir and Hubei, for SARS2, is no more remarkable than the distance travelled between Yunnan and Guangzhou for SARS1.
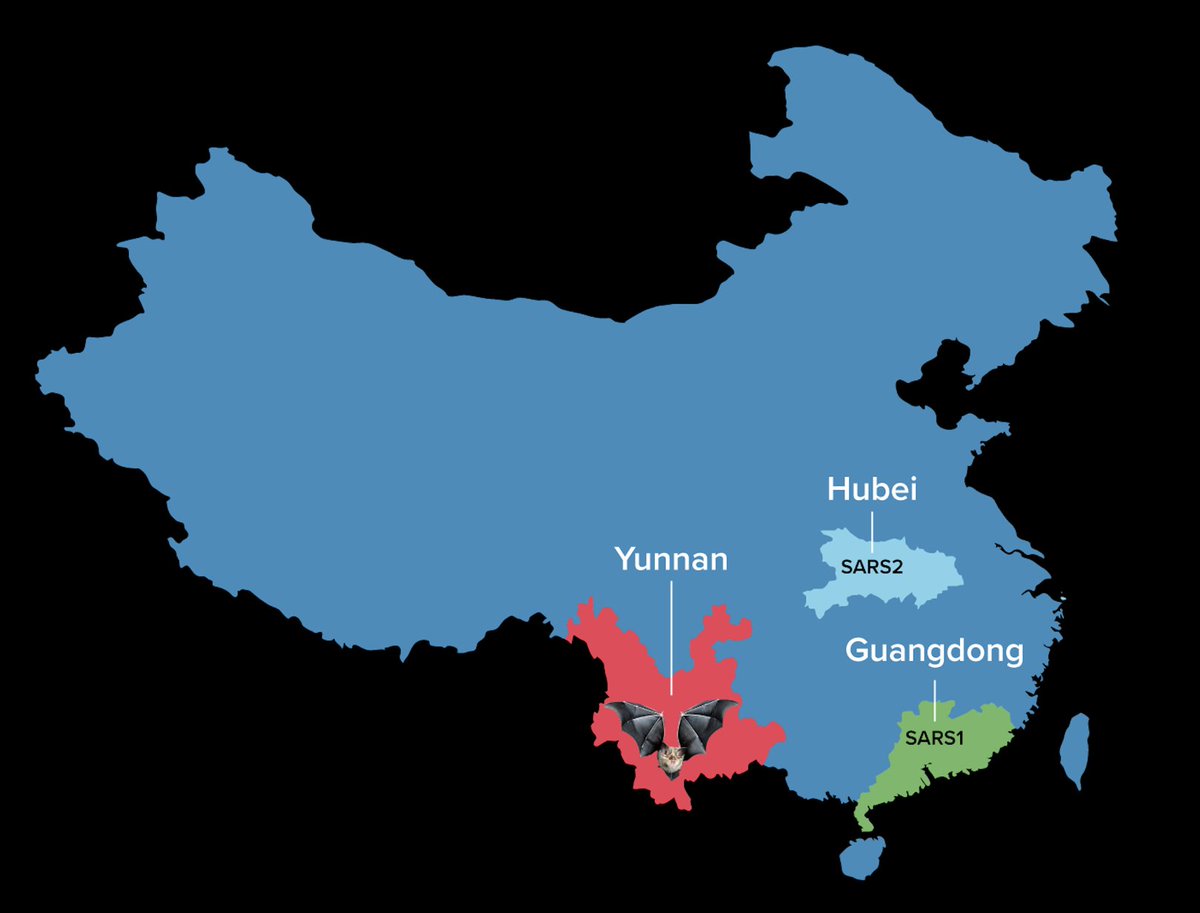
7/16 No sensible person claims on these grounds that SARS1 was due to a lab leak. So judge this argument for the origin of SARS2 - and the people promoting it - with that in mind.
Big h/t to @MilesFujimoto for the figure above and all the following graphics.
Big h/t to @MilesFujimoto for the figure above and all the following graphics.


8/16 Second, there is the assertion that it would be a crazy coincidence if SARS2 had emerged in Wuhan, of all places, if it *hadn't* come from a lab.
It is true that the Wuhan Institute of Virology is a major center of coronavirus study.
It is true that the Wuhan Institute of Virology is a major center of coronavirus study.
9/16 I can identify with anyone who thinks this is a crazy coincidence. I never quite thought it was crazy, but at one time I thought it was very provocative. But two things I've learned about this have altered my views completely.
10/16 The first is that SARS2 wasn't going to emerge just anywhere in China. Research I helped conduct, led by Joel Wertheim and @jepekar, showed that about 99% of the time a virus with the transmission properties of early SARS2 would jump into a human...
science.org/doi/10.1126/sc…
science.org/doi/10.1126/sc…
11/16 in a rural location, it would fizzle out. It really takes a city to allow a virus like this to take hold.

12/16 And, cities in China with a lab (or even 3 or 4 labs), which you could point to after the fact as the source of a pandemic that emerged there, are a dime a dozen.
Here's a map of (by one count) mainland China's ten most populous cities.
Here's a map of (by one count) mainland China's ten most populous cities.
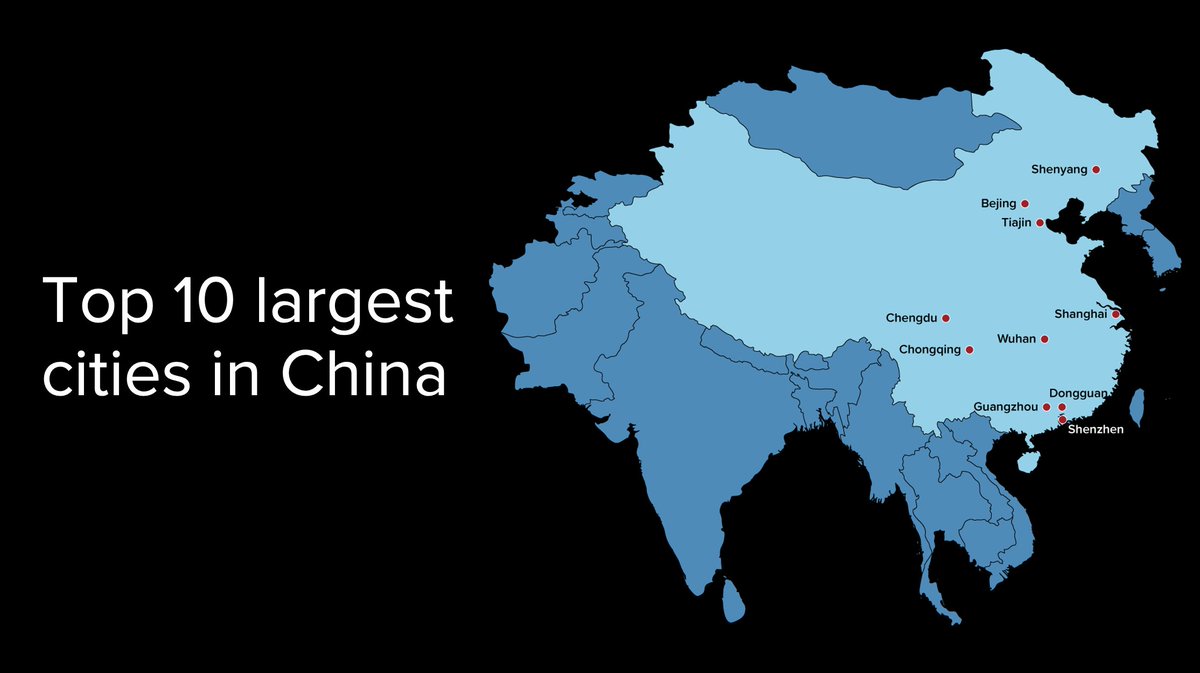
13/16 Turns out most major cities (Hong Kong too) have sites that could & would be blamed, after the fact, for a pandemic. There's even a lab in Beijing that collected bat virus samples from the 'Mojiang mine' and worked with @PeterDaszak and EcoHealth Alliance. A few examples:
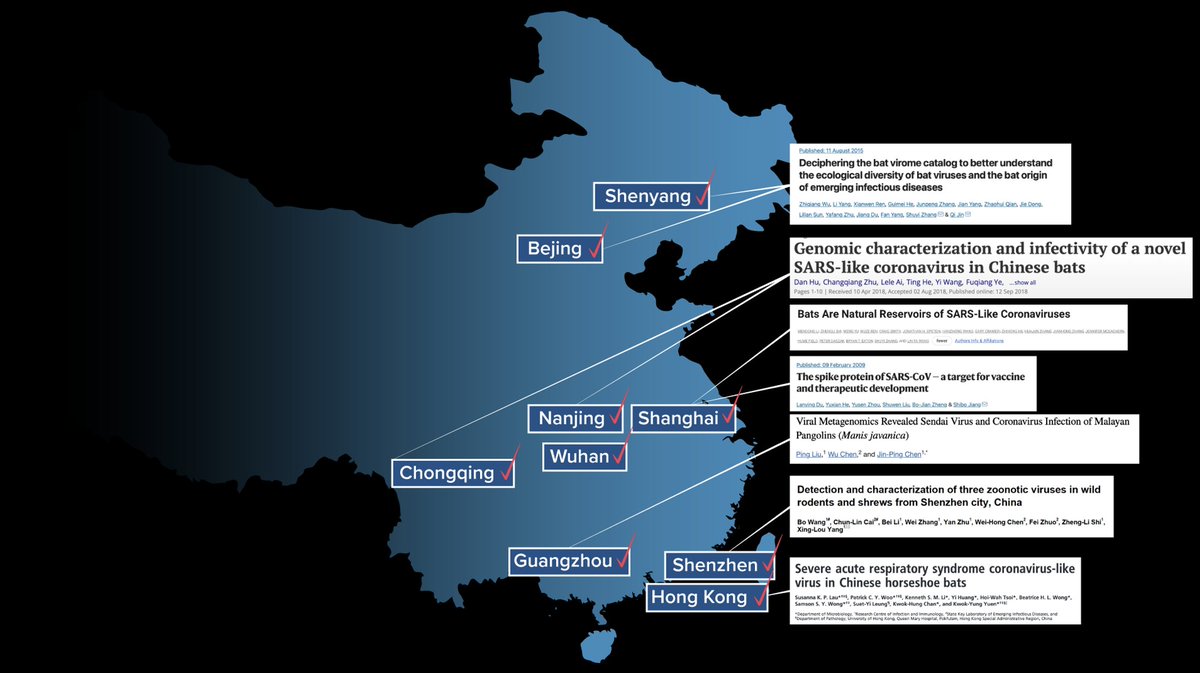
14/16 I think that many of us have fallen into a cognitive trap by positing that an outbreak in Wuhan - whose epicenter was some 15 km from the Wuhan Institute of Virology - started at the WIV...because otherwise there would be some crazy coincidence to explain.
15/16 Perfectly fine idea. But one that kind of falls apart when new analyses, and knowledge beyond the spotlight on the WIV, are taken into consideration.
16/16 It's a shame, but understandable, that Christopher Wray, the FBI, the 'Z Division' and the DOE, among others, appear not to understand this.


*Guangdong, I meant.
• • •
Missing some Tweet in this thread? You can try to
force a refresh