In new study, I have analyzed correlation between SARS-CoV-2 & animal genetic material in full set of environmental samples from Huanan Seafood Market.
Analysis clarifies what sequencing these samples can & cannot tell us about early outbreak at market.biorxiv.org/content/10.110…
Analysis clarifies what sequencing these samples can & cannot tell us about early outbreak at market.biorxiv.org/content/10.110…
Background:
China first reported coronavirus cases associated w market & no human transmission. But then we learned was human transmission & some early cases not from market.
Thus began still unanswered questions of role of market, nicely summarized here science.org/content/articl…
China first reported coronavirus cases associated w market & no human transmission. But then we learned was human transmission & some early cases not from market.
Thus began still unanswered questions of role of market, nicely summarized here science.org/content/articl…
In 2022, Chinese CDC released pre-print describing sampling market beginning Jan-1-2020:
They collected 457 animal & 923 environmental samples. They stated all animal samples tested negative, but 73 environmental samples positive.researchsquare.com/article/rs-137…
They collected 457 animal & 923 environmental samples. They stated all animal samples tested negative, but 73 environmental samples positive.researchsquare.com/article/rs-137…
In their 2022 pre-print, Chinese CDC described deep sequencing >150 environmental samples.
They included plot (below) that showed SARS-CoV-2 content of samples was correlated w human genetic material. From this, they concluded humans were source of virus in samples at market.
They included plot (below) that showed SARS-CoV-2 content of samples was correlated w human genetic material. From this, they concluded humans were source of virus in samples at market.

But Chinese CDC did not label other points on plot, so it wasn’t clear what other species had genetic material that correlated with SARS-CoV-2 abundance. They also didn’t provide raw sequencing data to enable other scientists to do this analysis.
This omission was widely noted in multiple news articles where scientists requested access to raw data to analyze which species correlated w SARS-CoV-2 ( & ).science.org/content/articl…
https://twitter.com/sciencecohen/status/1498658469775298568
Eventually, Chinese CDC uploaded some of the raw sequencing files to GISAID, where they were downloaded by another group of scientists who started analyzing data.
Before any written analysis posted, media started reporting that data suggested raccoon dogs may have been infected at market because their genetic material was co-mingled w SARS-CoV-2 in environmental samples ( & )nytimes.com/2023/03/16/sci…
theatlantic.com/science/archiv…
theatlantic.com/science/archiv…
The next week, the scientists reported their initial analysis, @critschristoph et al, of environmental samples:
Crits-Christoph et al reported some samples contained genetic material from raccoon dogs & other susceptible animal species (like bamboo rats).zenodo.org/record/7754299…
Crits-Christoph et al reported some samples contained genetic material from raccoon dogs & other susceptible animal species (like bamboo rats).zenodo.org/record/7754299…
Analysis by Crits-Christoph et al therefore genetically confirmed prior reports that species like raccoon dogs & bamboo rats were present at market.
The genetic details could inform tracing supply of these animals, which is important to investigate.
The genetic details could inform tracing supply of these animals, which is important to investigate.
However, Crits-Christoph et al did not report analysis of SARS-CoV-2 content of samples: they just used Chinese CDC classification of whether samples were “positive”.
So their analysis did not identify which species have genetic material correlated w viral material.
So their analysis did not identify which species have genetic material correlated w viral material.
Next week the Chinese CDC uploaded revised version of their pre-print, which shortly thereafter was published in Nature (). They also made all raw sequencing data available on public databases like SRA and NGDC.nature.com/articles/s4158…
New Chinese CDC paper agreed some samples had material from raccoon dogs & other species, although they did metagenomics differently than Crits-Christoph et al (probably not as well).
But they also did not analyze correlation of SARS-CoV-2 & animal genetic material in samples.
But they also did not analyze correlation of SARS-CoV-2 & animal genetic material in samples.
In fact, Chinese CDC even removed their earlier incompletely labeled SARS-CoV-2 vs species correlation plot that started all the questions.
So there still hasn’t been any analysis of how SARS-CoV-2 genetic material correlates w that of other animals!
So there still hasn’t been any analysis of how SARS-CoV-2 genetic material correlates w that of other animals!
https://twitter.com/sciencecohen/status/1498658469775298568
My new analysis addresses what animal genetic material correlates w SARS-CoV-2.
To do this, I wrote fully reproducible computational pipeline that downloads all raw sequencing data (which exceeds 3 terabytes) from NGDC database: github.com/jbloom/Huanan_…
To do this, I wrote fully reproducible computational pipeline that downloads all raw sequencing data (which exceeds 3 terabytes) from NGDC database: github.com/jbloom/Huanan_…
First, I confirmed data Chinese CDC uploaded to NGDC is superset of data analyzed by Crits-Christoph.
See SHA-512 file hashes:
So regardless of earlier controversy about data access, unmodified versions of all files now publicly available.github.com/jbloom/Huanan_…
See SHA-512 file hashes:
So regardless of earlier controversy about data access, unmodified versions of all files now publicly available.github.com/jbloom/Huanan_…
I then aligned sequencing reads to concatenated reference of SARS-CoV-2 & chordate mitochondrial genomes.
This enabled me to quantify both how much SARS-CoV-2 & mitochondrial genetic material from each species is in each sample.
This enabled me to quantify both how much SARS-CoV-2 & mitochondrial genetic material from each species is in each sample.
I correlated species compositions from my analysis with compositions reported by Crits-Christoph et al (which are only for mammals).
Results highly correlated (see figure below). This is good: two independent analyses get similar results for species compositions of samples.
Results highly correlated (see figure below). This is good: two independent analyses get similar results for species compositions of samples.

But note composition depends on reference set. I use chordates; Crits-Christoph et al report compositions for mammals.
Below are mitochondrial compositions for sample Q61: raccoon dog most abundant mammal, duck most abundant chordate.
Below are mitochondrial compositions for sample Q61: raccoon dog most abundant mammal, duck most abundant chordate.

Different if you align contigs to full genomes: then more raccoon dog than duck (Fig 1B of my pre-print)
There isn’t one correct way. Here I use mitochondrial composition to be consistent w Crits-Christoph et al report & because some species don’t have full genomes available.
There isn’t one correct way. Here I use mitochondrial composition to be consistent w Crits-Christoph et al report & because some species don’t have full genomes available.
You can go to to interactively look up mitochondrial species composition for any sample.jbloom.github.io/Huanan_market_…
Now for new part: what is SARS-CoV-2 content of samples?
Below is plot of percent of reads mapping to SARS-CoV-2 for each sample.
Most samples have little or no SARS-CoV-2. Samples with most SARS-CoV-2 have mitochondrial material mostly from fish.
Below is plot of percent of reads mapping to SARS-CoV-2 for each sample.
Most samples have little or no SARS-CoV-2. Samples with most SARS-CoV-2 have mitochondrial material mostly from fish.
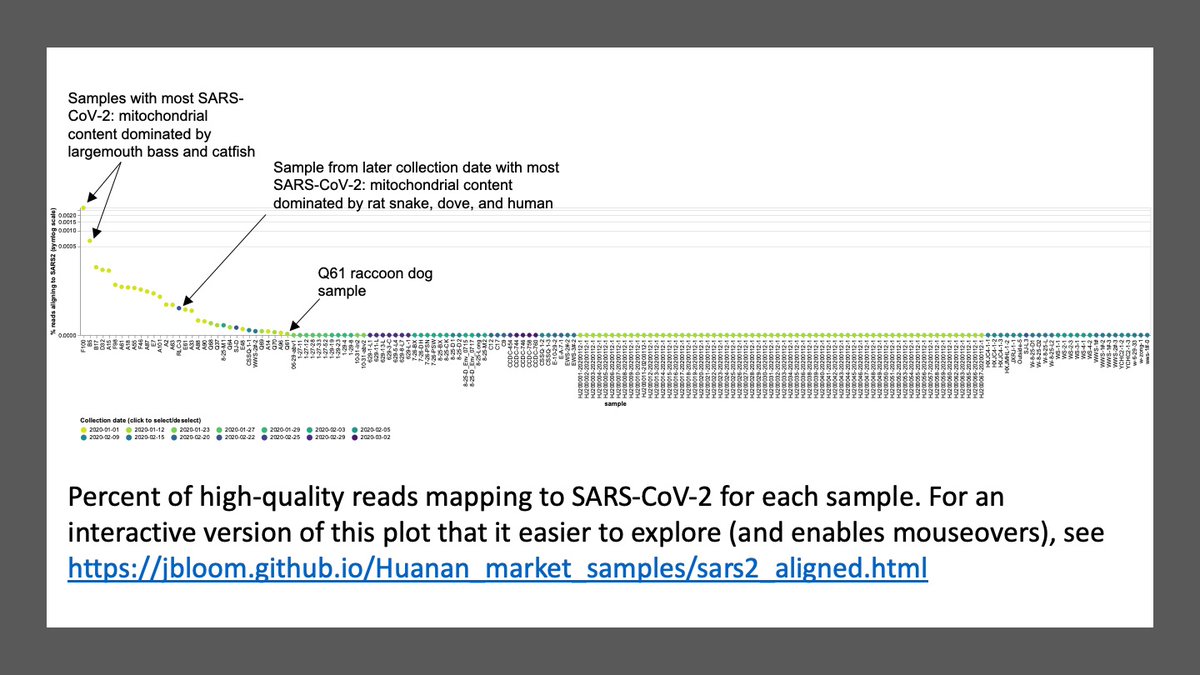
Many ways to partition samples: date collected, etc. Interactive plot at allows you to do that.
Eg, if we only look at later sampling dates, sample w most SARS-CoV-2 dominated by rat snake, dove, & human mitochondrial material.jbloom.github.io/Huanan_market_…
Eg, if we only look at later sampling dates, sample w most SARS-CoV-2 dominated by rat snake, dove, & human mitochondrial material.jbloom.github.io/Huanan_market_…
What about samples w lots of mitochondrial material from susceptible non-human animals like raccoon dogs?
Below is table of SARS-CoV-2 content of all samples with >20% of their chordate mitochondrial material from a susceptible non-human species.
Below is table of SARS-CoV-2 content of all samples with >20% of their chordate mitochondrial material from a susceptible non-human species.

There are 14 samples w >20% chordate mitochondrial material from raccoon dog: 13 have no SARS2 reads, other has 1 in ~200,000,000 reads mapping to SARS2
0 of 6 samples w >20% bamboo rat material have SARS2 reads
1 sample each w Malayan porcupine & Amur hedgehog have SARS2 reads
0 of 6 samples w >20% bamboo rat material have SARS2 reads
1 sample each w Malayan porcupine & Amur hedgehog have SARS2 reads
We can correlate number of SARS-CoV-2 reads w mitochondrial reads for each species across all samples (below).
Highest correlation for largemouth bass, catfish, cow, carp, snakehead fish
Humans modestly correlated w SARS2 reads
Raccoon dogs negatively correlated w SARS2 reads
Highest correlation for largemouth bass, catfish, cow, carp, snakehead fish
Humans modestly correlated w SARS2 reads
Raccoon dogs negatively correlated w SARS2 reads

There are many ways to subset data on sampling dates, calculate correlation, etc.
Interactive plots at & let you see how correlations change when you do that.jbloom.github.io/Huanan_market_…
jbloom.github.io/Huanan_market_…
Interactive plots at & let you see how correlations change when you do that.jbloom.github.io/Huanan_market_…
jbloom.github.io/Huanan_market_…
Finally, we can circle back to question scientists asked when Chinese CDC first posted pre-print in 2022: what if you label other species in correlation plot?
Below is that plot, shown only for samples w at least one SARS2 read for consistency w original Chinese CDC figure.
Below is that plot, shown only for samples w at least one SARS2 read for consistency w original Chinese CDC figure.
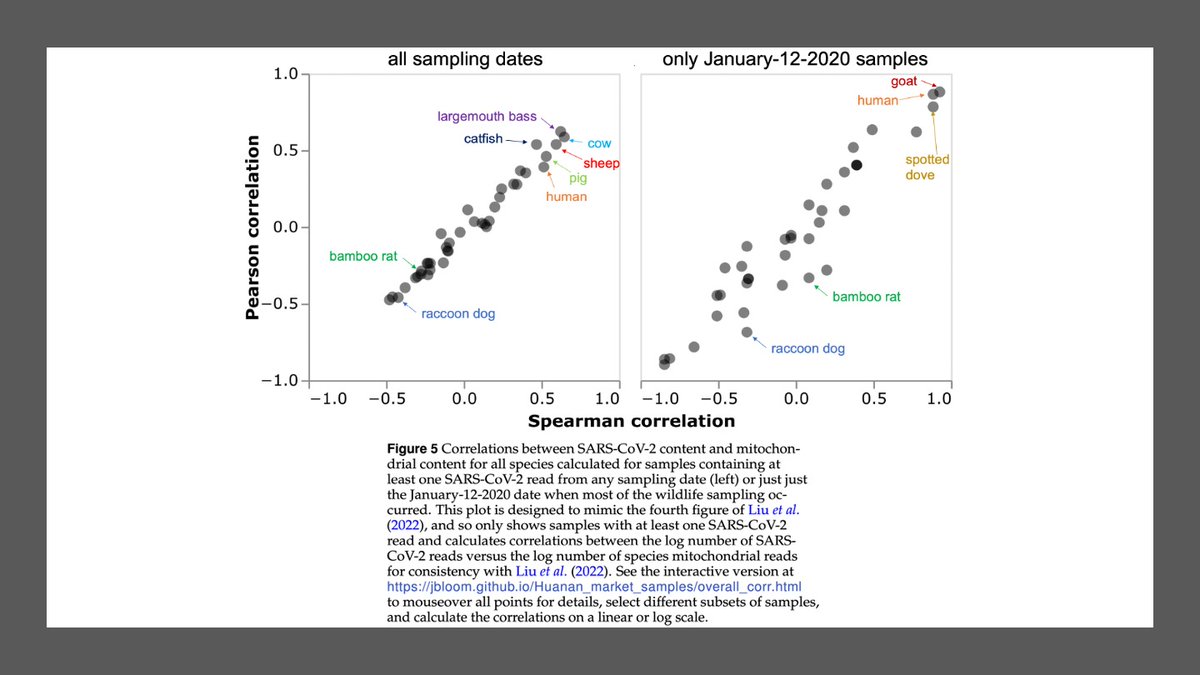
Species most correlated w SARS2 are fish & livestock, followed by humans. Raccoon dogs & bamboo rats negatively correlated w SARS2.
Similar if we only look at samples collected on Jan-12-2020, which was date of most intense wildlife stall sampling.
Similar if we only look at samples collected on Jan-12-2020, which was date of most intense wildlife stall sampling.
Again, lots of ways to subset samples & calculate correlations, & you can explore them using the interactive plots at jbloom.github.io/Huanan_market_…
So how did we end up w media articles about raccoon dog material co-mingled w SARS2?
Raccoon dogs are one of species least co-mingled w SARS2, and Q61 raccoon dog sample only has 1 of 200,000,000 reads mapping to SARS2.
May have to do w how Chinese CDC called sample positivity
Raccoon dogs are one of species least co-mingled w SARS2, and Q61 raccoon dog sample only has 1 of 200,000,000 reads mapping to SARS2.
May have to do w how Chinese CDC called sample positivity

In their pre-print/paper, Chinese CDC called “positive” any sample that either tested positive by RT-qPCR or had >0 sequencing reads mapping to SARS-CoV-2.
But these environmental samples: they mix various animal and/or viral sequences (plus probably index hopping in sequencing)
But these environmental samples: they mix various animal and/or viral sequences (plus probably index hopping in sequencing)
Criteria Chinese CDC used to call positivity aren’t consistent.
Eg, Q61 was negative by RT-qPCR, but has 1 of 200,000,000 reads mapping to SARS2. Not consistent to call Q61 positive but call negative other samples that also tested negative by RT-qPCR & were never sequenced.
Eg, Q61 was negative by RT-qPCR, but has 1 of 200,000,000 reads mapping to SARS2. Not consistent to call Q61 positive but call negative other samples that also tested negative by RT-qPCR & were never sequenced.

Therefore, I suggest future work should stop using “positive” / “negative” classification of Chinese CDC table, & instead analyze quantitative SARS2 content only across samples subjected to same consistent set of assays (eg, Ct values or SARS2 read content).
More broadly, what can we conclude about COVID-19 origins from all this?
Probably not much.
@DrTedros of @WHO had correct interpretation: we should analyze everything, but these data don’t tell us how pandemic began
Probably not much.
@DrTedros of @WHO had correct interpretation: we should analyze everything, but these data don’t tell us how pandemic began
https://twitter.com/WHO/status/1636704883091857409
Recall market samples were collected on Jan-1-2020 or later.
First human SARS2 infections in Wuhan occurred no later than Nov 2019.
By Jan 2022, SARS2 had been spread widely across market by humans, regardless of how it originated.
First human SARS2 infections in Wuhan occurred no later than Nov 2019.
By Jan 2022, SARS2 had been spread widely across market by humans, regardless of how it originated.
Viral material is most co-mingled w material from fish & livestock products, but virus clearly did NOT originate w those species & products.
It’s simply that environmental samples taken over month after humans started spreading virus do not reliably indicate outbreak origin.
It’s simply that environmental samples taken over month after humans started spreading virus do not reliably indicate outbreak origin.
If we ever learn origin of SARS2, I suspect it will come from information on events that occurred in Nov 2019 (or earlier):
Until then, we should analyze all available data--but be circumspect & cognizant of limits of these data & our knowledge.
Until then, we should analyze all available data--but be circumspect & cognizant of limits of these data & our knowledge.
https://twitter.com/jbloom_lab/status/1462231909430267906
Finally, interactive versions of all plots from my analysis are at
Computer code is at
Pre-print is at
I hope others explore & build on this computer code w further analyses.jbloom.github.io/Huanan_market_…
github.com/jbloom/Huanan_…
biorxiv.org/content/10.110…
Computer code is at
Pre-print is at
I hope others explore & build on this computer code w further analyses.jbloom.github.io/Huanan_market_…
github.com/jbloom/Huanan_…
biorxiv.org/content/10.110…
In response to discussion (), I don't think analysis disproves or proves source
Just emphasizes these samples collected too late to reveal origin
We need info on earlier events
Unless we get that, we need to acknowledge don't know exactly what happened
Just emphasizes these samples collected too late to reveal origin
We need info on earlier events
Unless we get that, we need to acknowledge don't know exactly what happened
https://twitter.com/profamirattaran/status/1651455986786525184
I was getting questions re 20% cutoff used to decide which samples to show in Table 1 of pre-print.
Analysis is of all samples, 20% cutoff is just to make Table 1 manageable in size.
For any sample w >1% chordate mitochondrial material from raccoon dogs, see bigger table below
Analysis is of all samples, 20% cutoff is just to make Table 1 manageable in size.
For any sample w >1% chordate mitochondrial material from raccoon dogs, see bigger table below

If you want SARS2 content of all samples ordered by raccoon dog mitochondrial %, see this bigger table:
If you want comparable data for all samples *and* all species, see this even bigger table: github.com/jbloom/Huanan_…
github.com/jbloom/Huanan_…
If you want comparable data for all samples *and* all species, see this even bigger table: github.com/jbloom/Huanan_…
github.com/jbloom/Huanan_…
However, these tables get so big they are difficult to look at.
That is point of interactive scatter plots here:
Choose any species, sampling date, etc & then see SARS2 vs mitochondrial content in one small plot & mouse over points for details.jbloom.github.io/Huanan_market_…
That is point of interactive scatter plots here:
Choose any species, sampling date, etc & then see SARS2 vs mitochondrial content in one small plot & mouse over points for details.jbloom.github.io/Huanan_market_…
I have posted updated version of preprint on bioRxiv:
This update includes addition of tables mentioned in three Tweets above, plus some revisions and responses to thoughtful comments by @flodebarre @acritschristoph detailed here:
biorxiv.org/content/10.110…
This update includes addition of tables mentioned in three Tweets above, plus some revisions and responses to thoughtful comments by @flodebarre @acritschristoph detailed here:
biorxiv.org/content/10.110…
https://twitter.com/jbloom_lab/status/1653933155445817346
The final peer-reviewed version of my analysis of the environmental samples at the Huanan market is now published in Virus Evolution: academic.oup.com/ve/article/9/2…
I have performed additional new analyses comparing SARSCoV2 to other animal CoVs in environmental samples from Huanan Market
Paper w these new analyses is & full computational pipeline at
The new results are summarized below.doi.org/10.1093/ve/vea…
github.com/jbloom/Huanan_…
Paper w these new analyses is & full computational pipeline at
The new results are summarized below.doi.org/10.1093/ve/vea…
github.com/jbloom/Huanan_…
I first calculated total reads mapping to SARSCoV2 & other animal CoVs across all samples, & just samples collected on date of wildlife stall sampling (Jan-12-2020)
Six CoVs have >500 reads; of these 4 have many reads from Jan-12-2020 samples, but 2 have few reads from that date
Six CoVs have >500 reads; of these 4 have many reads from Jan-12-2020 samples, but 2 have few reads from that date

Specifically, bamboo rat CoV, two canine CoVs, & rabbit CoV have substantial reads in samples from Jan-12-2020 wildlife-stall sampling
SARSCoV2 & rat CoV have few reads from that date
See for interactive plot w more options jbloom.github.io/Huanan_market_…
SARSCoV2 & rat CoV have few reads from that date
See for interactive plot w more options jbloom.github.io/Huanan_market_…

I next analyzed reads on per-sample basis. Below I just show results for Jan-12-2020 as that is when most samples w material from potentially susceptible animals (eg, raccoon dogs, bamboo rats) collected.

For bamboo rat, canine & rabbit CoVs there were samples w 100s viral reads. Samples w most viral reads had largest frac animal material from known hosts
But all Jan-12-2020 samples had few SARSCoV2 or rat CoV reads, & samples w most reads had little material from plausible hosts
But all Jan-12-2020 samples had few SARSCoV2 or rat CoV reads, & samples w most reads had little material from plausible hosts

To better explore the data in above plot, see for an interactive plot that enables you to mouseover points for details on samples, select individual CoVs to display, and show additional dates.jbloom.github.io/Huanan_market_…
I also plotted viral vs animal genetic content for Jan-12-2020 samples.
For 4 most abundant animal CoVs in these samples there is association of viral & host animal content, but not for much less abundant SARSCoV2 and rat CoV
(Also see interactive plot ) jbloom.github.io/Huanan_market_…
For 4 most abundant animal CoVs in these samples there is association of viral & host animal content, but not for much less abundant SARSCoV2 and rat CoV
(Also see interactive plot ) jbloom.github.io/Huanan_market_…

Overall, these results show that genetic material from some animal CoVs is fairly abundant in samples collected during the wildlife-stall sampling of the Huanan Market on Jan-12-2020. However, SARSCoV2 is not one of these CoVs.
For the animal CoVs with high abundance on Jan-12-2020, there are meaningful associations between the content of viral and animal genetic material. But there are not such associations for SARSCoV2 and less abundant viruses like the rat CoV Lucheng-19.
There remain significant caveats related to the underlying available data, as discussed in limitations section at end of my initial paper on this topic () academic.oup.com/ve/article/9/2…

Please see my full new paper at for more details, and interactive plots that allow you to explore the data in additional ways.doi.org/10.1093/ve/vea…
jbloom.github.io/Huanan_market_…
jbloom.github.io/Huanan_market_…
Corrected link to interactive plots: jbloom.github.io/Huanan_market_…
• • •
Missing some Tweet in this thread? You can try to
force a refresh