
1/9
CASE
Check out the CBC in the graphic below. Describe it in the fewest words possible.
CASE
Check out the CBC in the graphic below. Describe it in the fewest words possible.

2/9
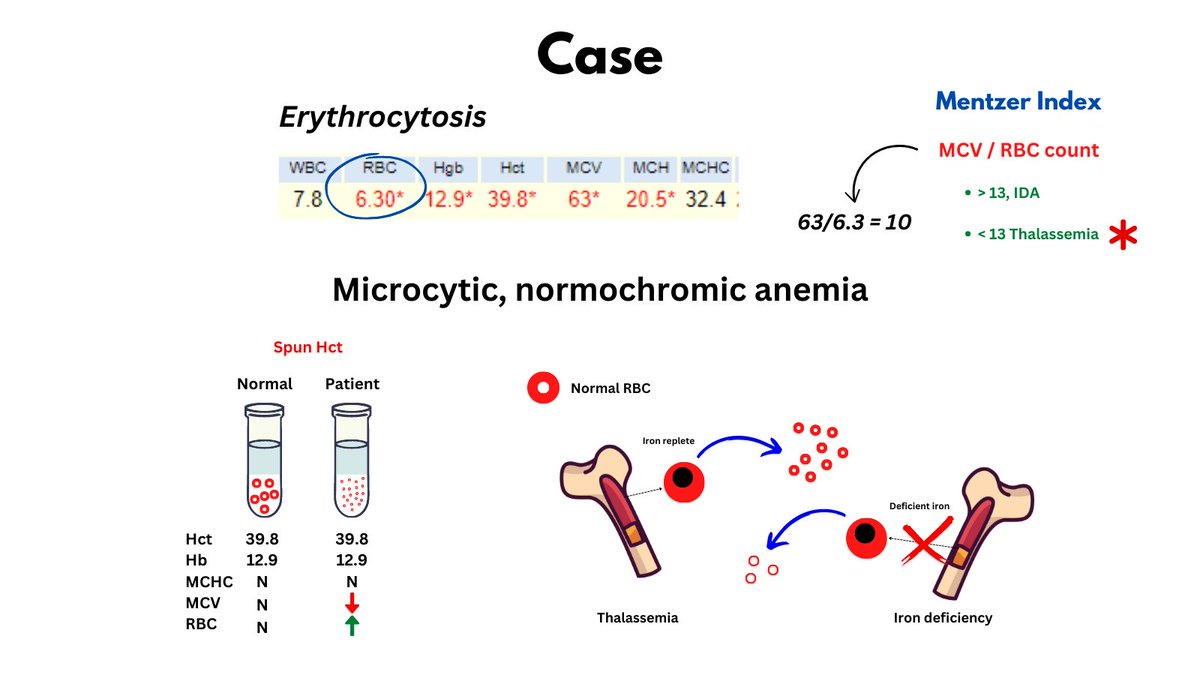
Normochromic microcytic anemia per Wintrobe's time-honored morphological classification of anemia (this patient is male, so has very mild anemia). May also describe the CBC as microcytic erythrocytosis. Likely diagnosis is thalassemia minor.
Normochromic microcytic anemia per Wintrobe's time-honored morphological classification of anemia (this patient is male, so has very mild anemia). May also describe the CBC as microcytic erythrocytosis. Likely diagnosis is thalassemia minor.

3/9
The numbers tell the story: lots of little red cells occupying a near normal fractional volume of blood on the account of increased red cell production by the bone marrow (Hct = RBC count x MCV), unimpeded by nutrient (i.e. Fe) deficiency.
The numbers tell the story: lots of little red cells occupying a near normal fractional volume of blood on the account of increased red cell production by the bone marrow (Hct = RBC count x MCV), unimpeded by nutrient (i.e. Fe) deficiency.
4/9
All data - so far - point to thalassemia minor. Now let's look at the patient's Hb electrophoresis (see graphic).
Note the increased HbA2 (c/w beta-thalassemia) and the massively increased HbF (normal < 1%).
All data - so far - point to thalassemia minor. Now let's look at the patient's Hb electrophoresis (see graphic).
Note the increased HbA2 (c/w beta-thalassemia) and the massively increased HbF (normal < 1%).

5/9
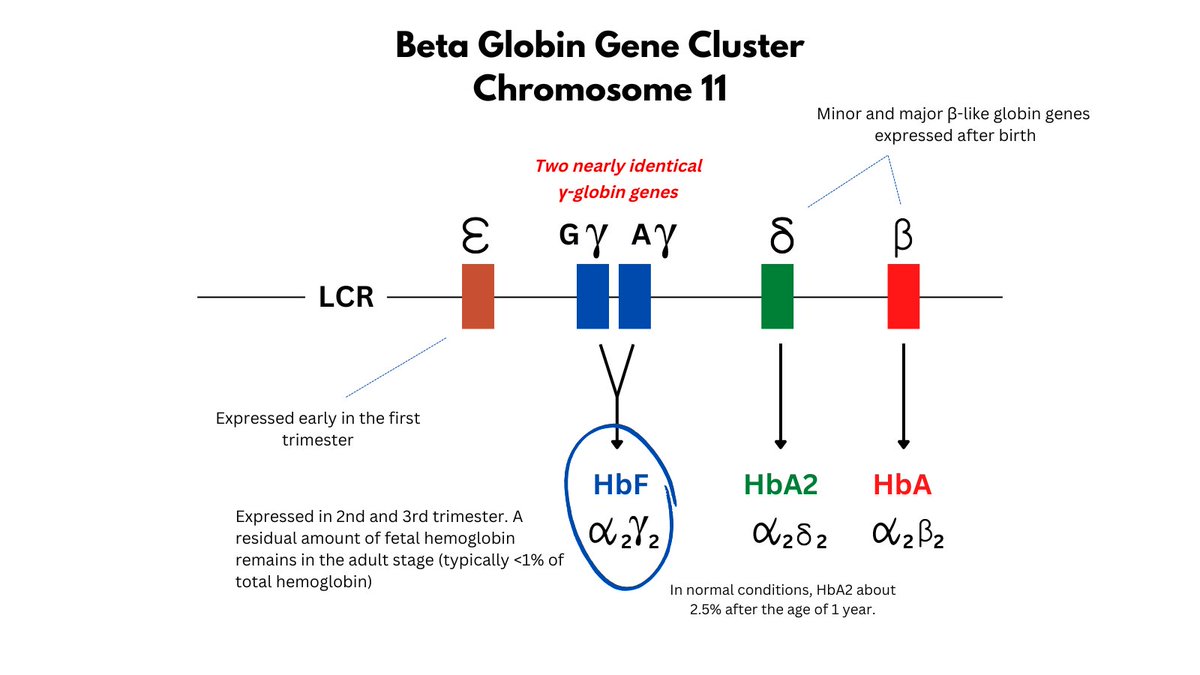
What is HbF?
It is a tetramer of 2 gamma (from the beta globin gene locus) and 2 alpha chains, expressed primarily in 2nd/3rd trimesters, but with low levels (< 1%) persisting in adults. There are actually 2 (very similar) genes that code for the gamma chain.
What is HbF?
It is a tetramer of 2 gamma (from the beta globin gene locus) and 2 alpha chains, expressed primarily in 2nd/3rd trimesters, but with low levels (< 1%) persisting in adults. There are actually 2 (very similar) genes that code for the gamma chain.
6/9
What causes elevated HbF levels? The causes can be grouped into hereditary and acquired (see graphic) (note: HbF levels in the normal population, while < 1%, actually vary a lot according to genetic factors).
What causes elevated HbF levels? The causes can be grouped into hereditary and acquired (see graphic) (note: HbF levels in the normal population, while < 1%, actually vary a lot according to genetic factors).

7/9
Different conditions lead to varying degrees of HbF elevation (see graphic). To determine the mechanism underlying this patient's remarkably high HbF, we carried out DNA sequencing. The patient was found to have a homozygous mutation in the promoter site of the beta gene.
Different conditions lead to varying degrees of HbF elevation (see graphic). To determine the mechanism underlying this patient's remarkably high HbF, we carried out DNA sequencing. The patient was found to have a homozygous mutation in the promoter site of the beta gene.

8/9
This is a nice example of how upregulation of the gamma chain can compensate for reduction in the beta chain in homozygous beta-thalassemia to maintain a near normal Hb.
This is a nice example of how upregulation of the gamma chain can compensate for reduction in the beta chain in homozygous beta-thalassemia to maintain a near normal Hb.
9/9
NOTE: HbF has a higher affinity for O2 compared with HbA (so the fetus can grab more O2 from mom), so this patient would be expected to have less O2 unloading in his tissues. This may provide an impetus for increased EPO secretion and thus a higher Hb.
NOTE: HbF has a higher affinity for O2 compared with HbA (so the fetus can grab more O2 from mom), so this patient would be expected to have less O2 unloading in his tissues. This may provide an impetus for increased EPO secretion and thus a higher Hb.
• • •
Missing some Tweet in this thread? You can try to
force a refresh















