
Now that I have a captive audience…(welcome new followers!) a monster thread on what my paper says, the approximations and the caveats… (1/aleph)
https://twitter.com/sineatrix/status/1686182852667572224
TLDR: My paper did *not* prove nor give evidence of superconductivity in Cu-apatite. It showed interesting structural and electronic properties that have features common with high-Tc superconductors provided Cu is in the right place. Otherwise we get a ‘boring’ semiconductor.
Firstly, it’s so exciting to see the interest in solid-state physics! Welcome to the most exciting subfield of physics ;) And thank you all for the interest!
Awesome to see the discussions and explanations of the work, especially those aimed at non-experts. I wish I could keep up and spend more time in these discussions but y’all broke my twitter. My personal fav is that I managed to see is from @MarjBaldwin
Now, down to it.
Last week when the experimental paper was announced I got plenty of messages....
Last week when the experimental paper was announced I got plenty of messages....
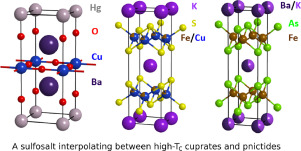
Looking at the structure, it didn’t look like something that would be high-Tc… why? A lot of the known high-TC materials are layered, and have substantial amounts of transition metals (e.g. Fe, Cu). This did not. Similar discussions on here including from @sentefmi
(Cuprates, left and pnictides, right -- known high-TC superconductors). Image from:
https://t.co/VKjIQPV0Sjsciencedirect.com/science/articl…
https://t.co/VKjIQPV0Sjsciencedirect.com/science/articl…
A detail in the paper that was interesting was the predicted volume collapse with Cu – I should be able to see that with the theoretical methods (more on that later) that I use, so I decided to start running the calculations.
Where to start? They had the chemical formula and space group in the paper (what the atoms are, how many there are, and where they go). I found it in the Materials Project. I also looked in a database for experimentally reported crystal structures in a database (ICSD).
I ran these two sets of calculations (one w/ O, one w/OH). Now that I had the structure, I had to decide which Pb atoms to replace with Cu. (Pink or Purple)
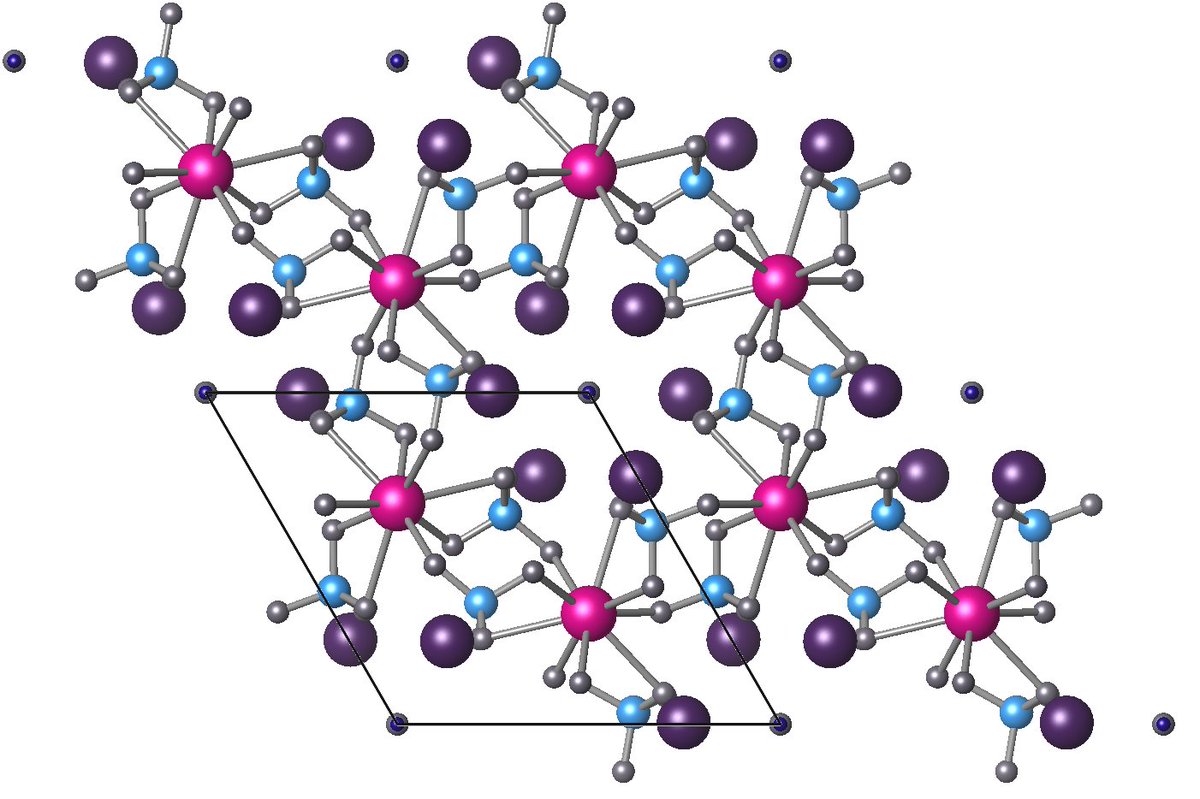
There are two inequivalent Pb atoms in the structure – I considered both (called Pb(1) and Pb(2)). They report to have Cu on Pb(1) in the exp (pink). Note: this is the opposite to the labeling in the exp paper however I adopted the conventional labeling used in other literature.
With the structures, the next was to perform calculations – here I used a standard method called Density Functional Theory (DFT) which uses approximations in practice to solve the quantum mechanics that governs the behavior of ions and electrons in a material.
It is extremely effective for a wide range of properties, but has its limitations. For one, the approximations made in the implementation of DFT do not deal with strong ‘electron-electron’ interactions well.
You can expect to see these el-el effects as you go further down the periodic table and are pushing more electrons into orbitals.
Does that mean DFT is rubbish for anything other than the top row of the periodic table? No – we have several ways of augmenting DFT to better include some of these electron-electron interactions.
One of these is called DFT+U where the U is added to the orbitals that we expect to be treated inadequately. In my case, we expect the d-orbitals in Cu need such a treatment. There are several ways of deciding what value of U should be added (e.g. comparing with experiment).
Here I looked at a few reasonable values of U to ensure that I was getting a good match with whatever exp data we had (structure), and that the physics we were focusing on didn’t vary much with this parameter.
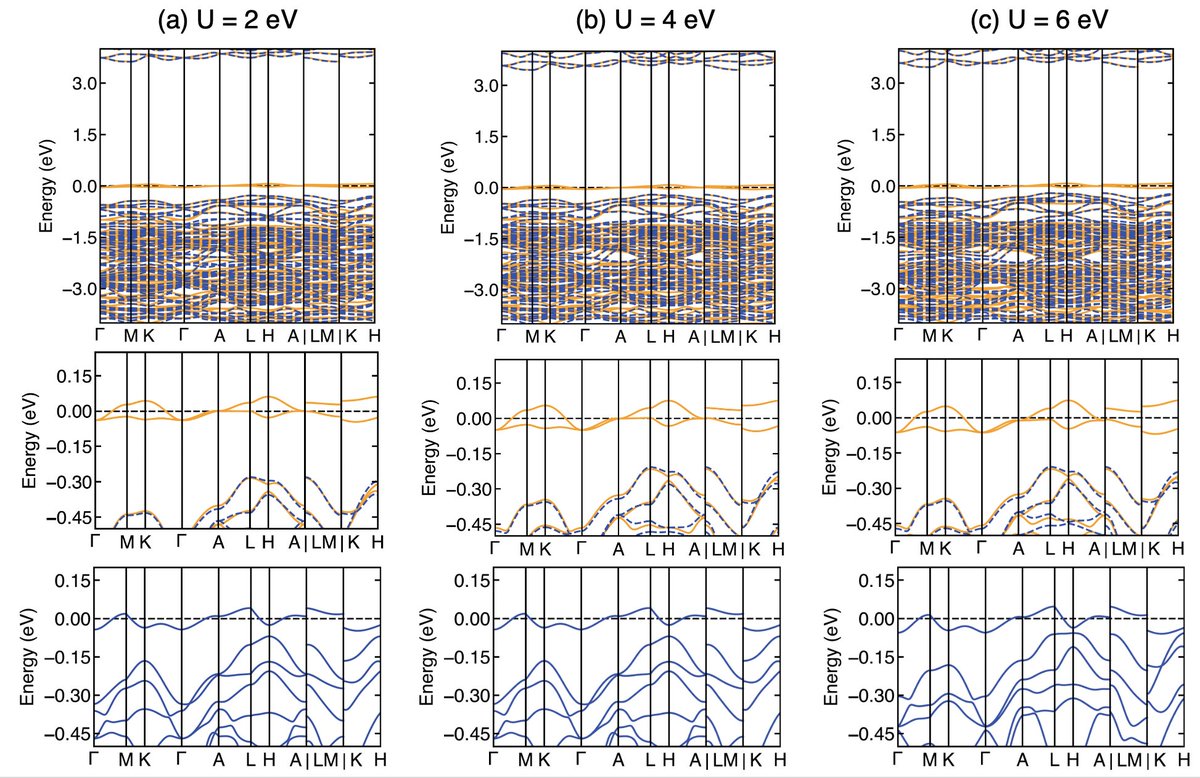
Result 1: Cu on the Pb(1) site causes a volume collapse. This is not especially exciting but was consistent with the experiment. However, what was particularly interesting to me was that the Cu substitution caused the whole crystal structure to collectively distort
(this is a bit weird for this small amount of substitution). I pulled a chemist colleague into my office to ask him if he also thought it was weird. He agreed.
Result 2: The electronic structure of Cu on Pb(1) has isolated flat bands at the Fermi level. These are a direct result of the structural distortion! When I calculate the bands without the distortion, they are not isolated. When I include the distortion, they are!
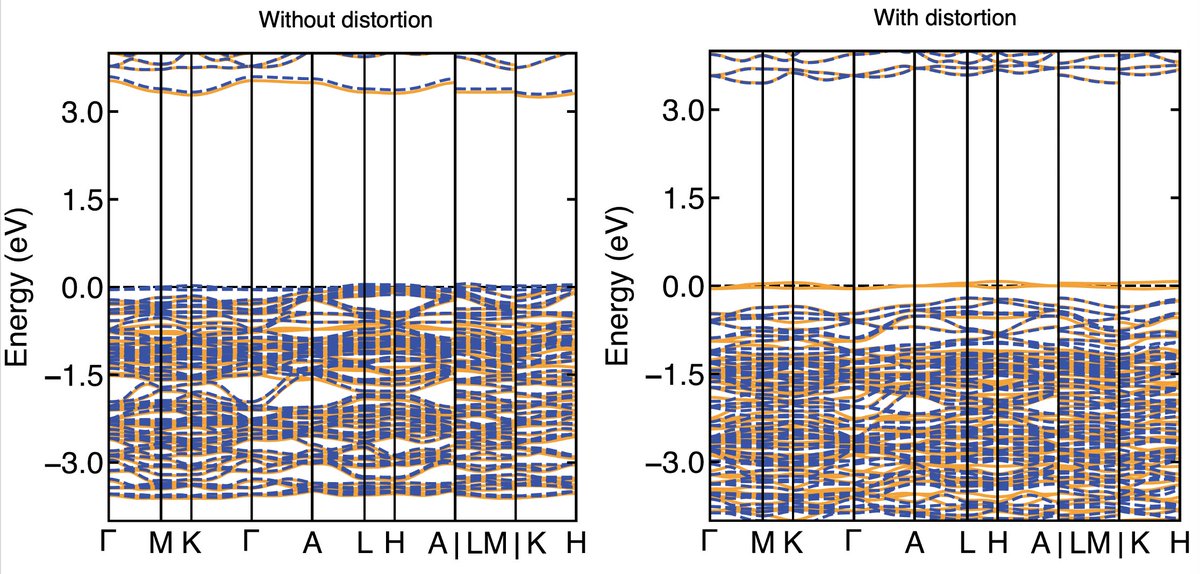
Result 3: The electronic structure of Cu on the Pb(2) does not have evidence of these flat bands and instead looks like a regular semiconductor. Moreover Cu is more stable on this Pb(2) site than on the Pb(1) site in the DFT.
What does this mean for the experiments?
What does this mean for the experiments?
If Cu is indeed sitting in the Pb(1) site in the apatite structure as they report, then the DFT calculations suggest that there are interesting flat bands that are a result of the structural distortion.
Flat bands can mean superconductivity, but can also mean a wealth of other phenomena like metal-insulator transitions, charge density waves, magnetism (all interesting!)
The structural distortion and my magnetic calculations of Cu on the Pb(1) also suggest that there might be strong electron-phonon coupling or spin fluctuations in this system (important for the superconducting mechanism)
END OF MEGATHREAD 1
END OF MEGATHREAD 1
• • •
Missing some Tweet in this thread? You can try to
force a refresh



