Q: What could GO WRONG with a major cancer genomics study in which tumours are sequenced to only 15x depth?
A: An awful LOT! 😟
Our detailed reanalysis now out in @RSocPublishing:
🧵👇 (1/18) tinyurl.com/RSOSrebuttal

A: An awful LOT! 😟
Our detailed reanalysis now out in @RSocPublishing:
🧵👇 (1/18) tinyurl.com/RSOSrebuttal

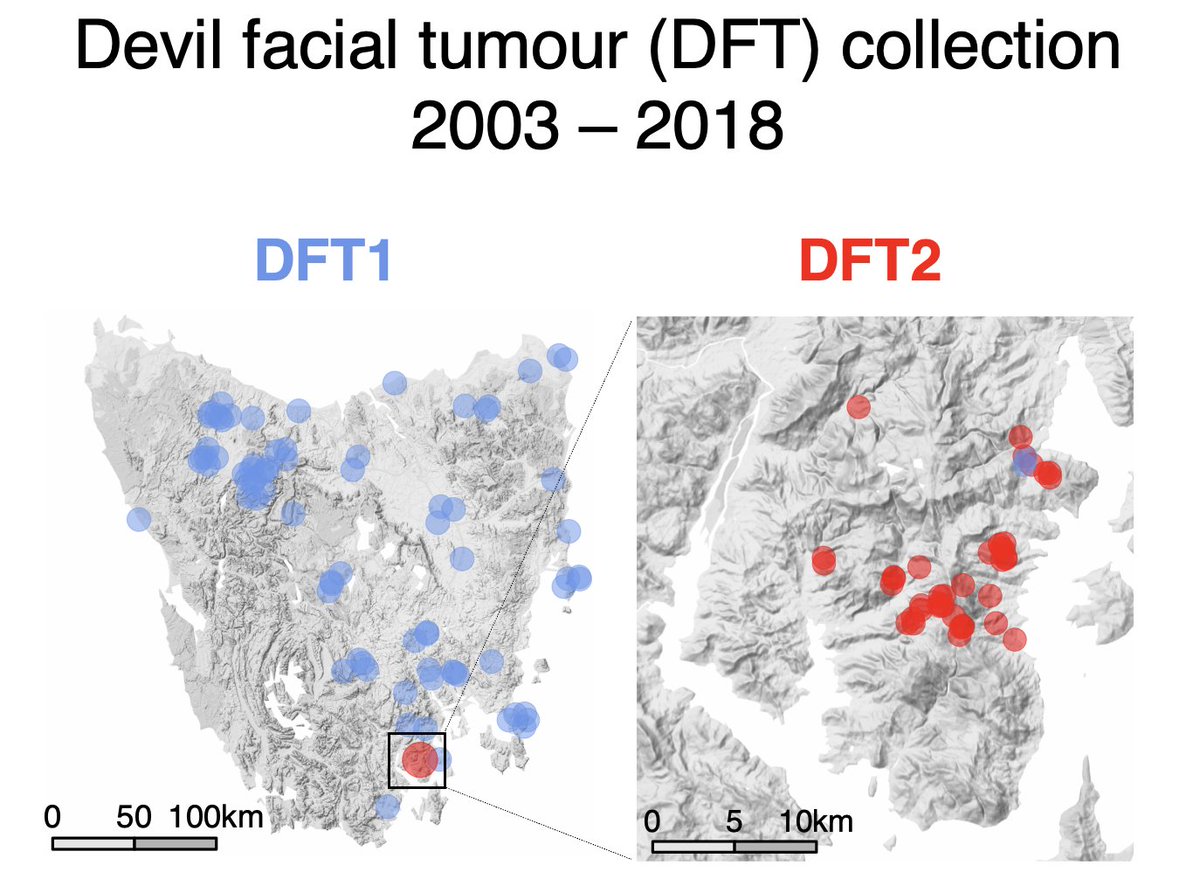
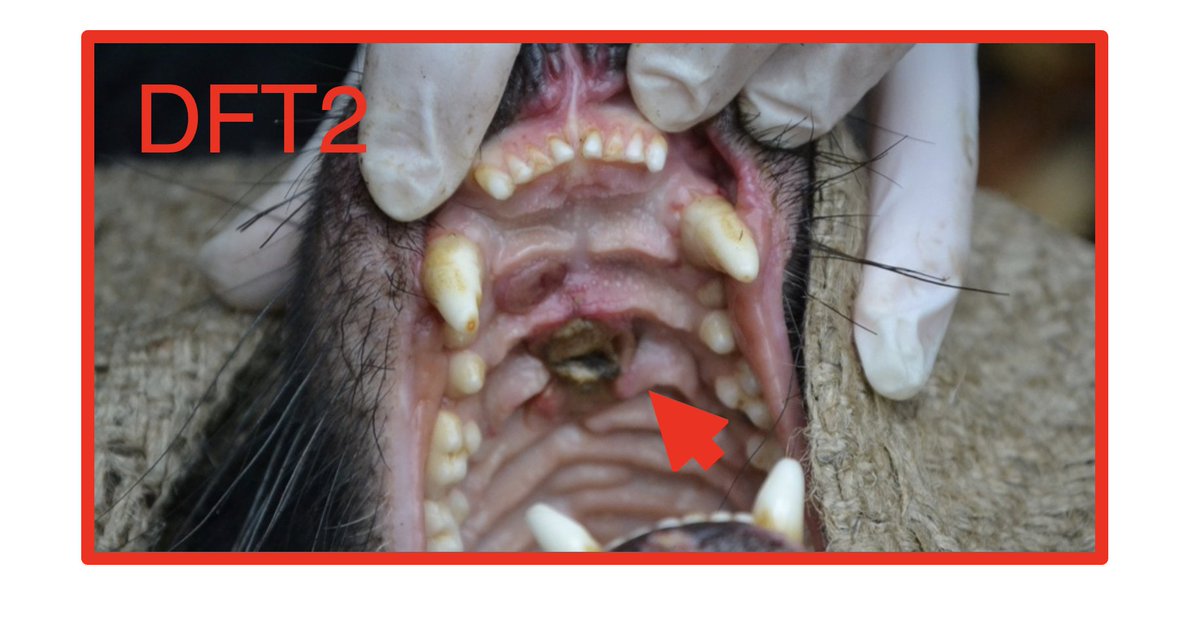
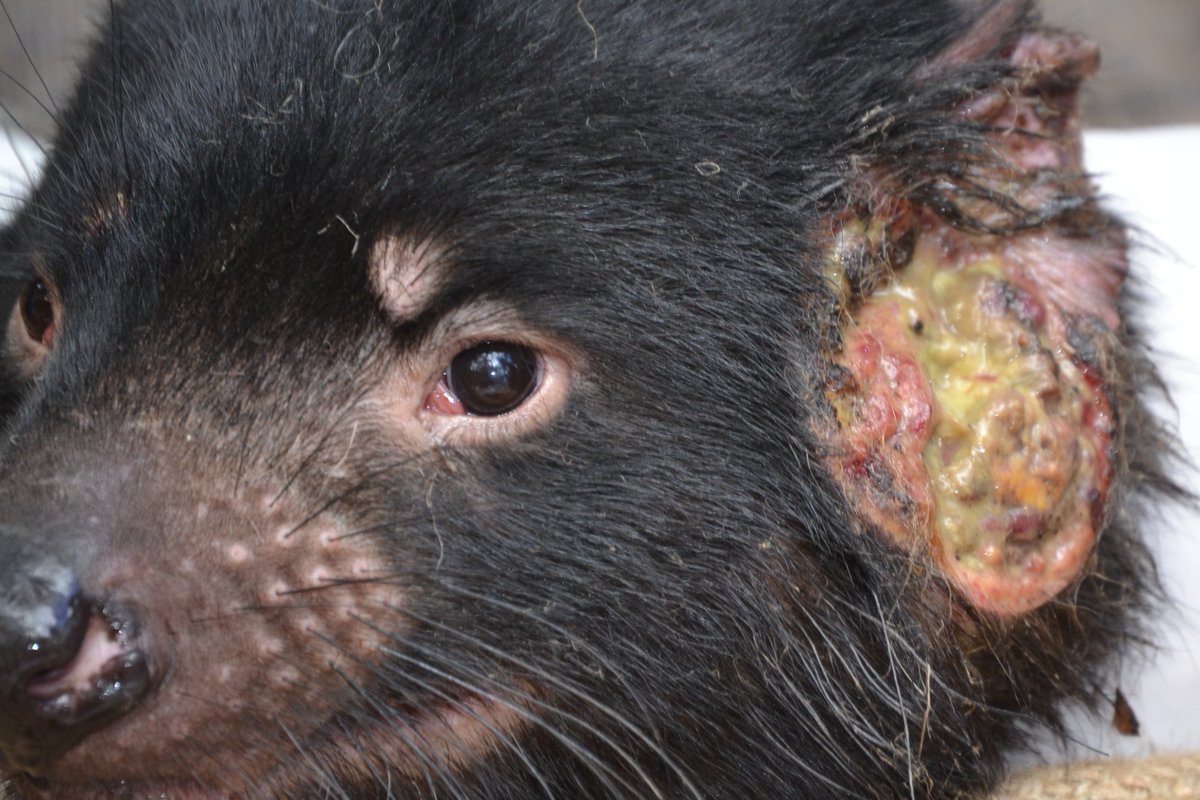
In 2020, the Storfer lab reported an analysis of the evolution of devil facial tumour disease () – a severe conservation threat.
Working on DFT, but coming to very different conclusions, we decided to dig deep into their DNA sequencing data. 🧑💻
🧵 (2/18) tinyurl.com/Tasdevils2020
Working on DFT, but coming to very different conclusions, we decided to dig deep into their DNA sequencing data. 🧑💻
🧵 (2/18) tinyurl.com/Tasdevils2020
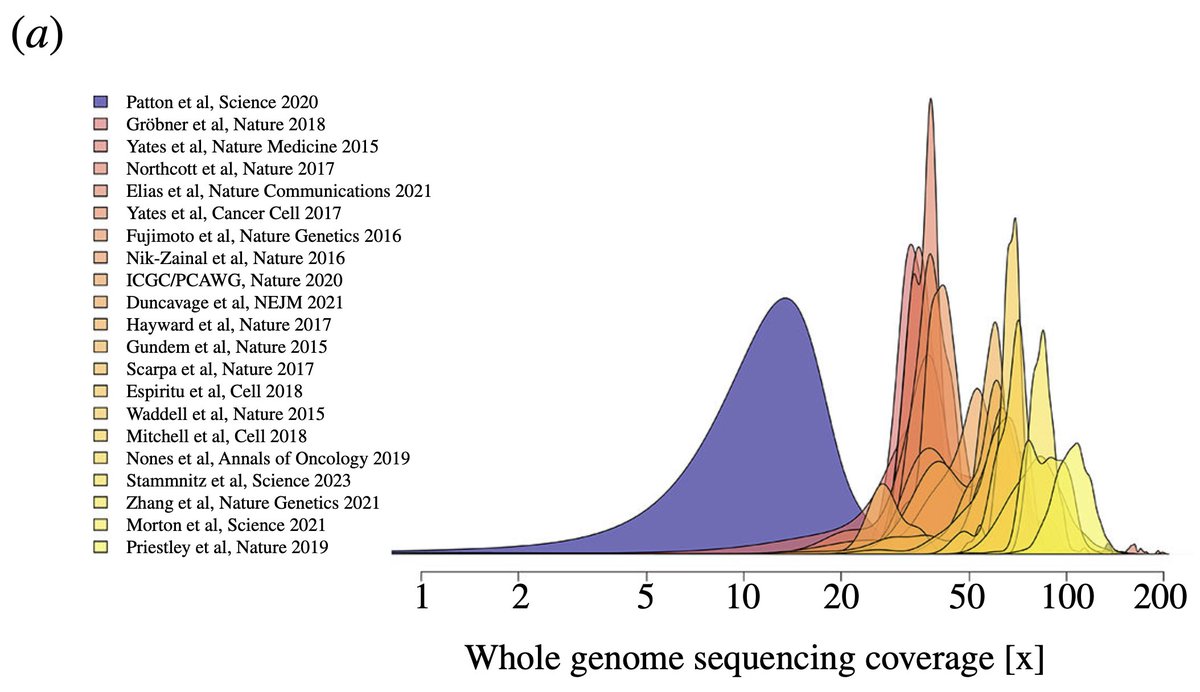
This plot shows whole genome sequencing depths across sample cohorts of twenty large-scale cancer genome data sets since 2015. A minimum of 30x is the standard, many modern studies reach >>60x.
In blue: the 51 devil tumours of the study in question – LOOKS LOW? 🪫🧬
🧵 (3/18)
In blue: the 51 devil tumours of the study in question – LOOKS LOW? 🪫🧬
🧵 (3/18)
Using a shared 25 Mbp deletion in DFT1s, we calculated these samples’ TUMOUR PURITY – the actual fraction of cancer cell DNA captured in the biopsies:
11 out of 51 samples feature <30 % purity. Tumour-only WGS coverage thereby drops from a median of 15x to 9x. 🔬📉
🧵 (4/18)
11 out of 51 samples feature <30 % purity. Tumour-only WGS coverage thereby drops from a median of 15x to 9x. 🔬📉
🧵 (4/18)
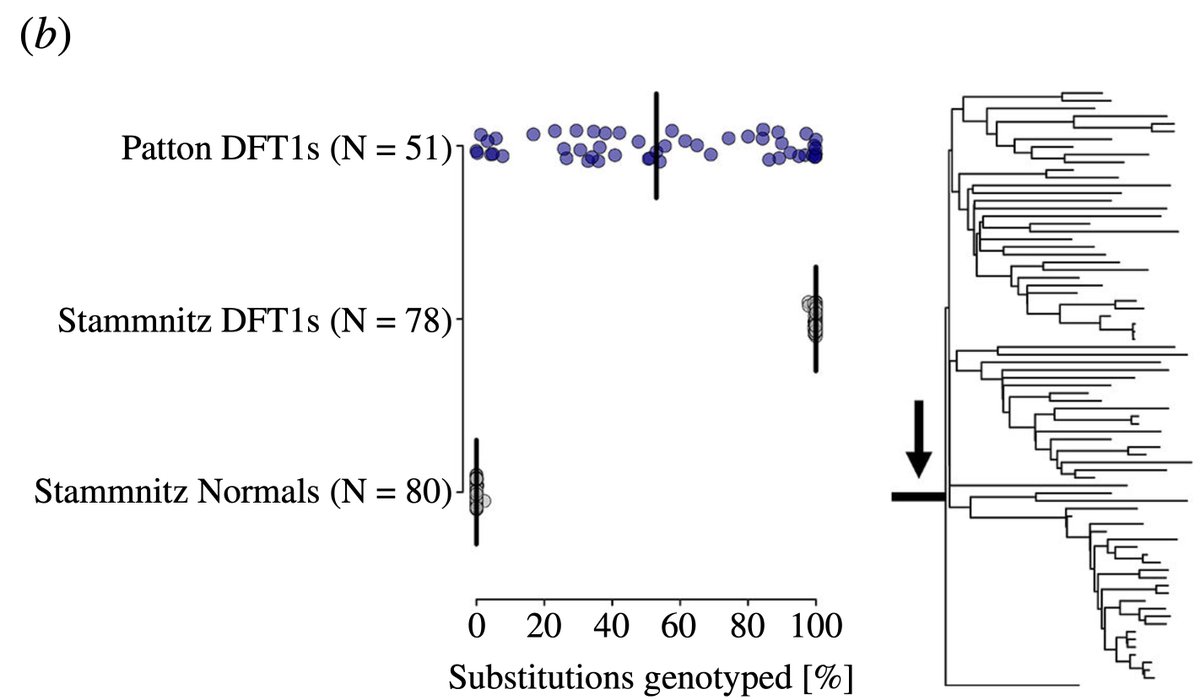
The consequence? UNRELIABLE MUTATION counts.
We genotyped ~1,300 point mutants which occurred early in the evolution of DFT1, are thus expected to be present in all tumours but absent from any normal devil:
On average, only 53 % (!) of substitutions are detected. 😐
🧵 (5/18)
We genotyped ~1,300 point mutants which occurred early in the evolution of DFT1, are thus expected to be present in all tumours but absent from any normal devil:
On average, only 53 % (!) of substitutions are detected. 😐
🧵 (5/18)
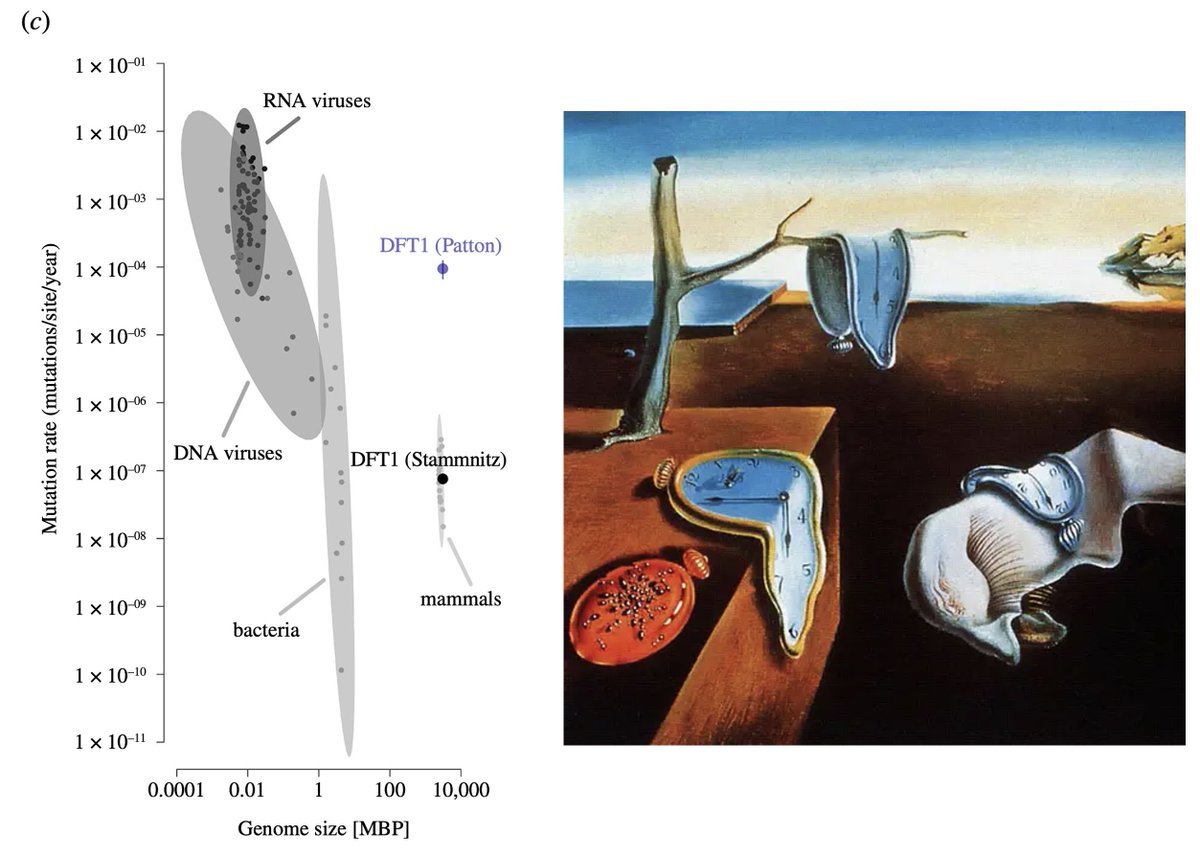
Half of the real DFT1 point mutations are missed.
And yet, using only a tiny interval of the entire devil genome, this study claims a MASSIVE MUTATION BURDEN:
2-3 orders of magnitude above mammalian rate estimates. How? Without evidence for a hypermutator process? 🤷♂️
🧵 (6/18)
And yet, using only a tiny interval of the entire devil genome, this study claims a MASSIVE MUTATION BURDEN:
2-3 orders of magnitude above mammalian rate estimates. How? Without evidence for a hypermutator process? 🤷♂️
🧵 (6/18)
This article presents a tree model fit, aiming to capture the evo-history of the epidemic. If you study DEVILS: 🚨
1. The DFT1 origin here is not in line with field observations, which point to northeastern Tasmania
2. Inferred DFT1 spread and migration 'jumps' don't make sense
1. The DFT1 origin here is not in line with field observations, which point to northeastern Tasmania
2. Inferred DFT1 spread and migration 'jumps' don't make sense
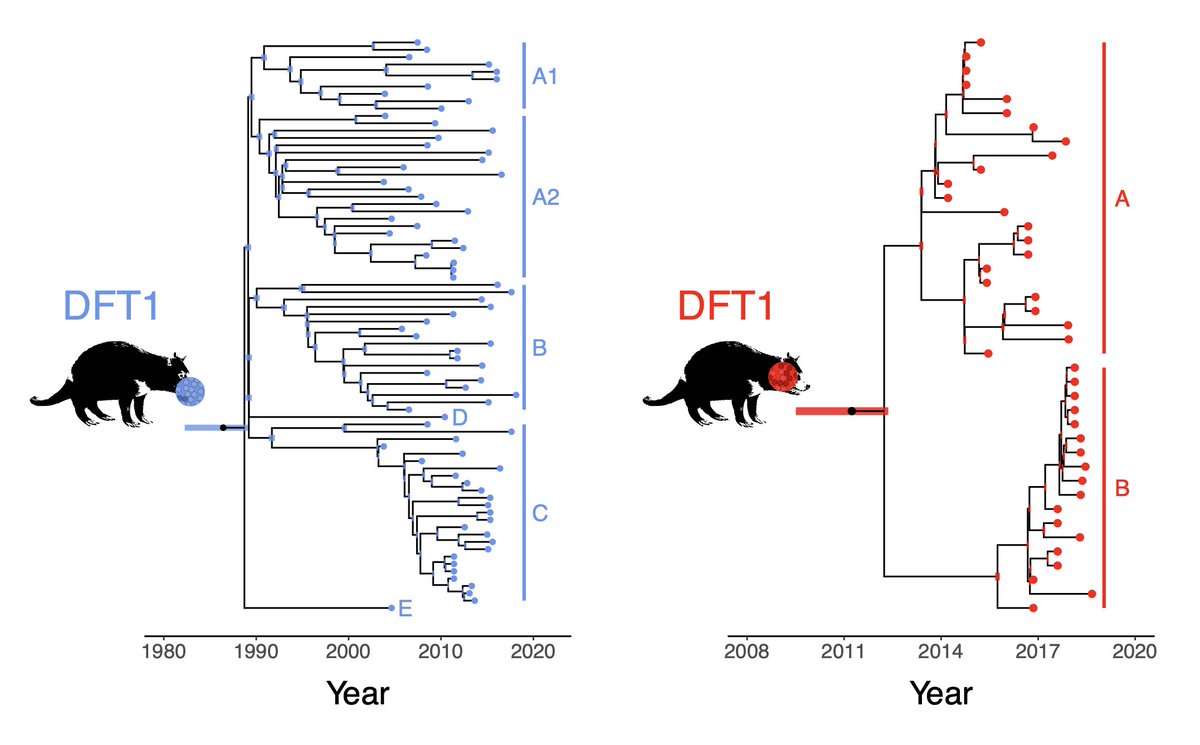
We decided to rebuild a more ACCURATE PHYLOGENY from their data.
Though rather than relying on point mutations from shallow sequencing, we focused on large chromosomal deletions and amplifications – which are reliable within 100 kb windows. 🔎🧬
And what happens? ...
🧵 (8/18)
Though rather than relying on point mutations from shallow sequencing, we focused on large chromosomal deletions and amplifications – which are reliable within 100 kb windows. 🔎🧬
And what happens? ...
🧵 (8/18)
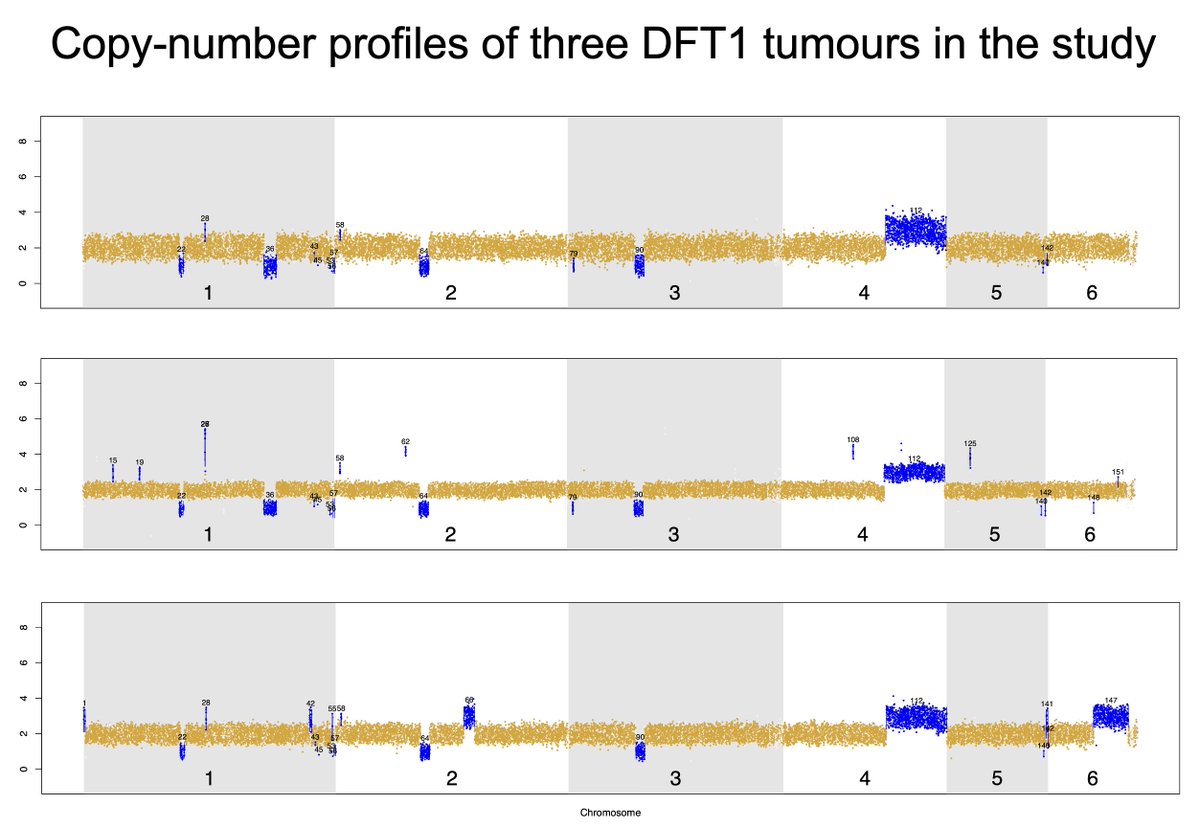
... the original study’s tree, based on noisy point mutations, and our large copy-number based model LOOK NOTHING ALIKE! It’s 🍏 vs 🍊!
Plot colours correspond to the four main DFT1 clades seen in our own data from >600 tumours – see Fig1 in .
🧵 (9/18) tinyurl.com/plosbio2020
Plot colours correspond to the four main DFT1 clades seen in our own data from >600 tumours – see Fig1 in .
🧵 (9/18) tinyurl.com/plosbio2020
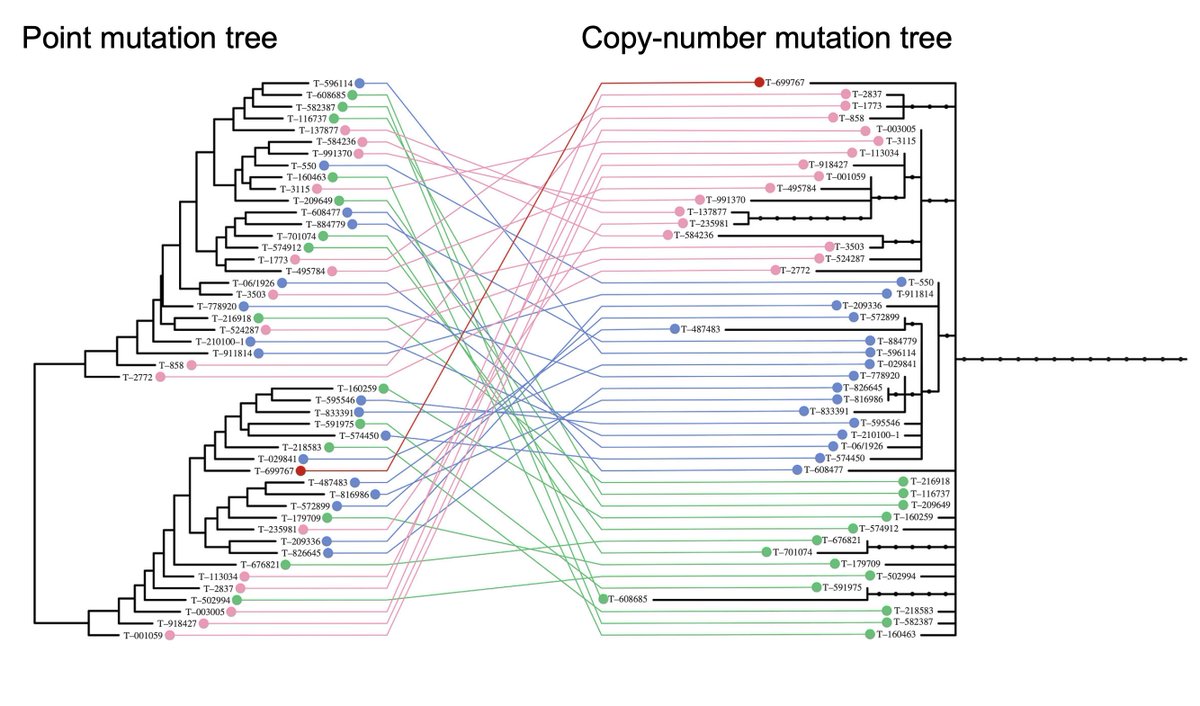
So very, very likely the DFT1 tree of this study has NO SCIENTIFIC BASIS.
With serious consequences! Because the paper's main conclusions and media hype, 'cautious optimism for the continued survival of the Tasmanian devil' are all derived from this flawed data. 😞
🧵(10/18)
With serious consequences! Because the paper's main conclusions and media hype, 'cautious optimism for the continued survival of the Tasmanian devil' are all derived from this flawed data. 😞
🧵(10/18)

There are MORE ISSUES with this study, only to mention a few:
- disregard for tumour purity, ploidy and clonality assumptions
- SNP filtering 'panel' of only 12 animals
- list of 'identified' somatic mutations not available
- final tumour WGS seq. depths unmentioned
🧵 (11/18)
- disregard for tumour purity, ploidy and clonality assumptions
- SNP filtering 'panel' of only 12 animals
- list of 'identified' somatic mutations not available
- final tumour WGS seq. depths unmentioned
🧵 (11/18)
How could this have been avoided? Three key data QC concepts in (cancer) genomics:
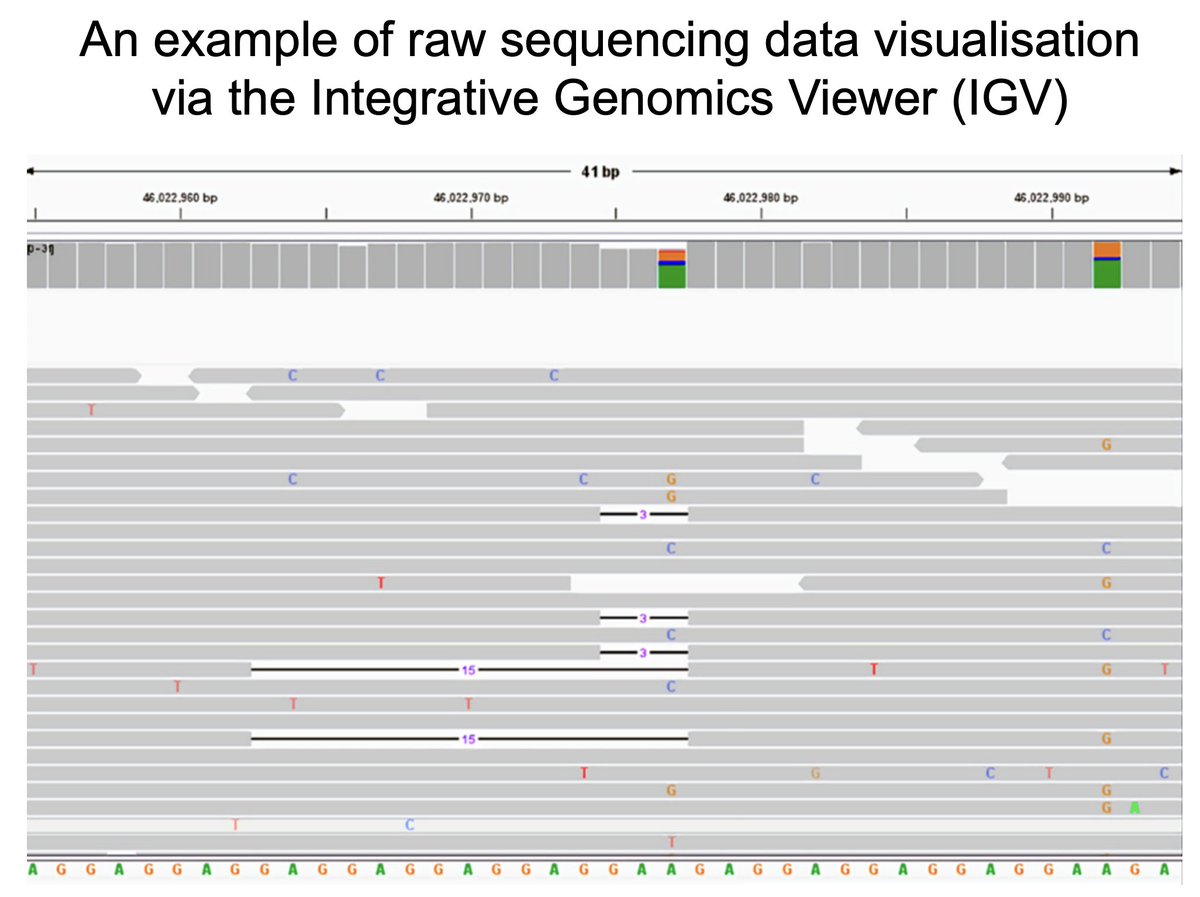
#1: RAW DATA visualisation of sequence alignments against the reference genome. 🔎🧬
Calibrate sensitive strategies and filters to distinguish real (somatic) mutations from noise.
🧵 (12/18)
#1: RAW DATA visualisation of sequence alignments against the reference genome. 🔎🧬
Calibrate sensitive strategies and filters to distinguish real (somatic) mutations from noise.
🧵 (12/18)
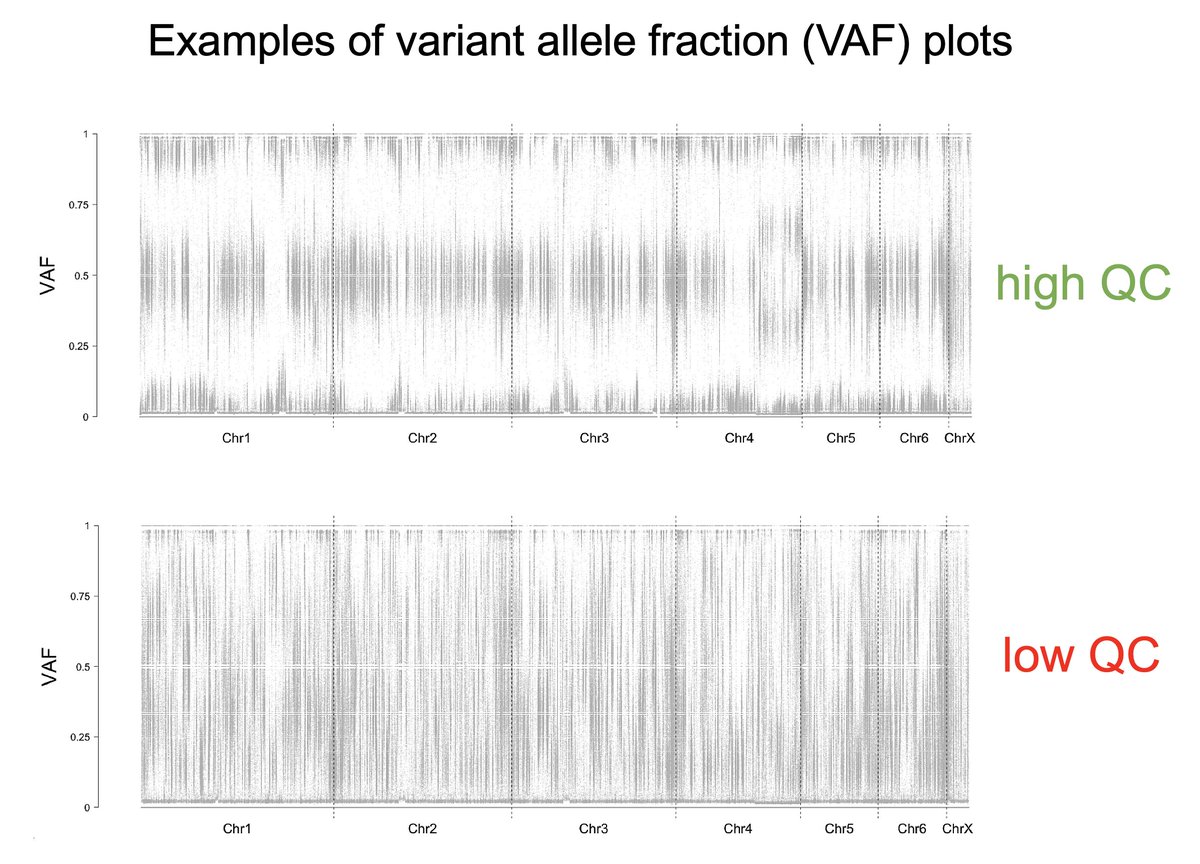
#2: VARIANT ALLELE FRACTION (VAF) profiling of tumour genomes. 🔎🧬
One should expect a VAF peak at ~50% because the exact same mutation usually only hits one of the two alleles.
Peaks << 50% can indicate tumour impurities; blurry spectra indicate sequencing noise.
🧵 (13/18)
One should expect a VAF peak at ~50% because the exact same mutation usually only hits one of the two alleles.
Peaks << 50% can indicate tumour impurities; blurry spectra indicate sequencing noise.
🧵 (13/18)
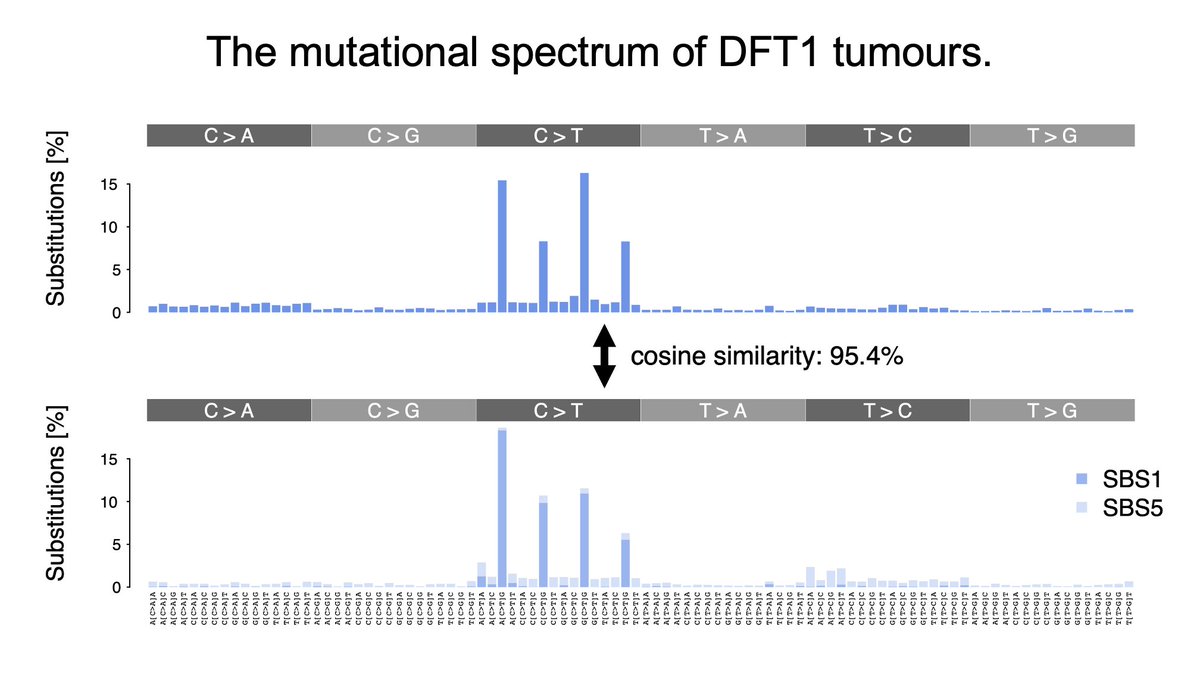
#3: MUTATIONAL SPECTRUM profiling of tumour genomes. 🔎🧬
DFT1 tumours mostly feature the widely known endogenous signatures SBS1 and SBS5, with characteristic peaks. Low-quality point mutation/substitution calls flag up in the spectra.
🧵 (14/18)
DFT1 tumours mostly feature the widely known endogenous signatures SBS1 and SBS5, with characteristic peaks. Low-quality point mutation/substitution calls flag up in the spectra.
🧵 (14/18)
On a broader note, our observations reminds me of other “spectacular” genomics studies in which the sequencing data were not treated adequately.
For example this recent re-analysis effort by @StevenSalzberg1’s lab @JohnsHopkins:
🧵 (15/18)
For example this recent re-analysis effort by @StevenSalzberg1’s lab @JohnsHopkins:
🧵 (15/18)
https://twitter.com/StevenSalzberg1/status/1711367878338125976
In the future, I hope that we can define better reporting standards for large-scale genomics projects.
Is it too much for journals and reviewers to ask for openly accessible summary lists of studies’ sample SEQUENCING METRICS, such as read coverage and mapping rates?
🧵 (16/18)
Is it too much for journals and reviewers to ask for openly accessible summary lists of studies’ sample SEQUENCING METRICS, such as read coverage and mapping rates?
🧵 (16/18)
I genuinely wish we never had to do this piece. It is painful to scrutinise and then criticise others’ work efforts to the high extent which we felt compelled to do here – especially when you value some of the devil researchers involved with the original study ☮️ ...
... though is there a better way than re-examining the actual research data, in-depth, to openly improve the scientific record?
The greater community perspective, assisting with the survival of this iconic marsupial, needs to stand above all personal interests. 🐾
🧵 (17/18)
The greater community perspective, assisting with the survival of this iconic marsupial, needs to stand above all personal interests. 🐾
🧵 (17/18)

This was a trio-effort with @kevin_gori and Liz Murchison @tcgcambridge, we wish to thank the @RSocPublishing #RoyalSocietyOpenScience editors and reviewers who commented on our reanalyses & carefully (re-)read the original devil cancer genome study. 📜
🧵 (18/18)
🧵 (18/18)
@ENASequence, @ensembl, @emblebi, @NCBI, @ERC_Research, @CRGenomica, @WCRIFoundation, @UofGIntegrity, @OSFramework, @ZENODO_ORG, @galaxyproject, @genomicsedu, @HHS_ORI, @Tagesspiegel, @MicrobiomDigest #reproducibility #researchintegrity
• • •
Missing some Tweet in this thread? You can try to
force a refresh