
My written testimony to Congress prior to testifying last week (full version with footnotes available at ):
Dear Chairman Massie,
Thank you for the invitation to testify before the House Judiciary Subcommittee ... in the hearing titled, Follow the Science?: Oversight of the Biden Covid-19 Administrative State Response.
Our firm’s vaccine practice, which spans vaccine injury, exemptions, and policy, has over forty professionals. In our lawsuits, we must prove claims regarding these products with government and high impact journal data and sources. Appeals to credentials do not suffice.
Please find below a few points regarding Covid-19 vaccines we believe provide a broader framework in which to consider the administrative state’s actions regarding these products. While these points may conflict with the cultural cognition of some, based on our experience, they reflect the best available evidence.judiciary.house.gov/sites/evo-subs…
Dear Chairman Massie,
Thank you for the invitation to testify before the House Judiciary Subcommittee ... in the hearing titled, Follow the Science?: Oversight of the Biden Covid-19 Administrative State Response.
Our firm’s vaccine practice, which spans vaccine injury, exemptions, and policy, has over forty professionals. In our lawsuits, we must prove claims regarding these products with government and high impact journal data and sources. Appeals to credentials do not suffice.
Please find below a few points regarding Covid-19 vaccines we believe provide a broader framework in which to consider the administrative state’s actions regarding these products. While these points may conflict with the cultural cognition of some, based on our experience, they reflect the best available evidence.judiciary.house.gov/sites/evo-subs…
1. Covid-19 Vaccine Trials Were Robust When Compared with Other Vaccine Trials
The pivotal trials relied upon to license Covid-19 vaccines were robust as compared to the trials relied upon to license most childhood vaccines.
The following chart compares the pivotal trials for Pfizer and Moderna’s Covid-19 vaccines with those for FDA licensed vaccines that CDC recommends be injected (three times each) between birth and six months of age:
The above data is easily confirmed by reviewing the source material on FDA’s website for each product. For example, below is a screenshot of Section 6.1 of the package insert for the Hep-B vaccine in the chart above:
The trial reports submitted to FDA to license this Hep-B vaccine (obtained via FOIA) also confirm it was licensed for infants based on a trial with 147 infants and children and 5 days of safety monitoring after injection. FDA is also over three years late in substantively responding to a petition regarding this patently invalid trial.
As another example, Prevnar 13 was licensed for babies based on a trial in which Prevnar was used as a control:
In turn, Prevnar was licensed based on a trial in which another experimental vaccine, an “Investigational meningococcal group C conjugate vaccine,” was used as a control:
A chart of each vaccine licensed by FDA on CDC’s childhood schedule, along with the control, safety review period, and link to FDA source for each, is available at . This chart reflects that none of the vaccines on CDC’s childhood schedule were licensed by FDA based on a long-term placebo-controlled trial and most were licensed based on days or weeks of safety follow up after injection. Hence, in comparison with the trials relied upon to license childhood vaccines, the trials for Covid-19 vaccines were robust.
(See Section 2 in next tweet below)icandecide.org/no-placebo
The pivotal trials relied upon to license Covid-19 vaccines were robust as compared to the trials relied upon to license most childhood vaccines.
The following chart compares the pivotal trials for Pfizer and Moderna’s Covid-19 vaccines with those for FDA licensed vaccines that CDC recommends be injected (three times each) between birth and six months of age:
The above data is easily confirmed by reviewing the source material on FDA’s website for each product. For example, below is a screenshot of Section 6.1 of the package insert for the Hep-B vaccine in the chart above:
The trial reports submitted to FDA to license this Hep-B vaccine (obtained via FOIA) also confirm it was licensed for infants based on a trial with 147 infants and children and 5 days of safety monitoring after injection. FDA is also over three years late in substantively responding to a petition regarding this patently invalid trial.
As another example, Prevnar 13 was licensed for babies based on a trial in which Prevnar was used as a control:
In turn, Prevnar was licensed based on a trial in which another experimental vaccine, an “Investigational meningococcal group C conjugate vaccine,” was used as a control:
A chart of each vaccine licensed by FDA on CDC’s childhood schedule, along with the control, safety review period, and link to FDA source for each, is available at . This chart reflects that none of the vaccines on CDC’s childhood schedule were licensed by FDA based on a long-term placebo-controlled trial and most were licensed based on days or weeks of safety follow up after injection. Hence, in comparison with the trials relied upon to license childhood vaccines, the trials for Covid-19 vaccines were robust.
(See Section 2 in next tweet below)icandecide.org/no-placebo




2.Covid-19 Vaccine Trials Anemic When Compared to Drug Trials
While Covid-19 vaccine trials were robust as compared to trials for vaccines on the CDC childhood schedule, they were anemic as compared to trials for most drugs.
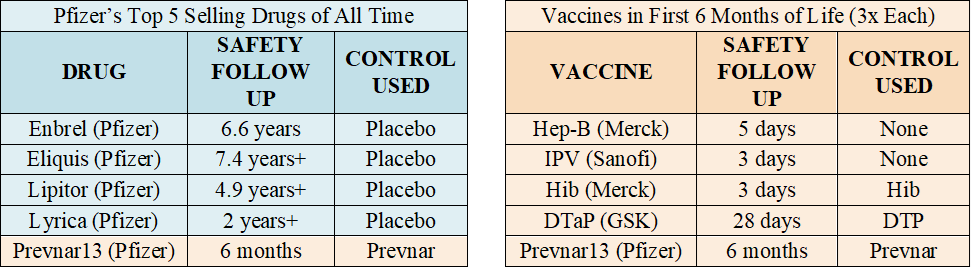
The table below (left) lists Pfizer’s top five selling products, according to one publication. The four drugs listed were each licensed based on a long-term placebo-controlled trial – the one vaccine listed, Prevnar 13, was not. The table below (right) again lists FDA licensed vaccines CDC recommends be injected (three times each) between birth and six months.
Clinical trials are critical for assuring safety. After a vaccine is licensed, it is considered unethical to conduct a placebo-controlled trial. Yet, while drugs are typically licensed based on long-term placebo-controlled trials, this is not true of FDA licensed vaccines on CDC’s childhood schedule. (It is also noted that when another vaccine was used as a control, that “control” vaccine was also not licensed based on a long-term placebo-controlled trial. )
This provides further context for the trials relied upon to license Covid-19 vaccines. While these trials were robust when compared to other vaccines, they are anemic when compared to trials relied upon to license drug products.
(See Section 3 in next tweet below)
While Covid-19 vaccine trials were robust as compared to trials for vaccines on the CDC childhood schedule, they were anemic as compared to trials for most drugs.
The table below (left) lists Pfizer’s top five selling products, according to one publication. The four drugs listed were each licensed based on a long-term placebo-controlled trial – the one vaccine listed, Prevnar 13, was not. The table below (right) again lists FDA licensed vaccines CDC recommends be injected (three times each) between birth and six months.
Clinical trials are critical for assuring safety. After a vaccine is licensed, it is considered unethical to conduct a placebo-controlled trial. Yet, while drugs are typically licensed based on long-term placebo-controlled trials, this is not true of FDA licensed vaccines on CDC’s childhood schedule. (It is also noted that when another vaccine was used as a control, that “control” vaccine was also not licensed based on a long-term placebo-controlled trial. )
This provides further context for the trials relied upon to license Covid-19 vaccines. While these trials were robust when compared to other vaccines, they are anemic when compared to trials relied upon to license drug products.
(See Section 3 in next tweet below)
3.Economic Interest to Assure Safety in Drug Trials Absent in Vaccine Trials
Economic interests that incentivize a company to assure the safety of its product before release into the market are absent for vaccine products.
Trials for drugs and vaccines are conducted by the pharmaceutical company seeking licensure. For drug products, companies remain liable for injuries the drugs cause after licensure. This provides an incentive to conduct long-term placebo-controlled trials to confirm the safety of drug products before licensure to avoid financial loss after licensure.
In contrast, for vaccine products, this economic incentive was eliminated by the National Childhood Vaccine Injury Act of 1986 (the “1986 Act”) which gave companies immunity for injuries caused by most vaccines. This is why long-term placebo-controlled trials for vaccine products do not make financial sense for companies seeking to maximize profits. In fact, while assuring safety in drug trials is aligned with a company’s economic interest, it is in conflict in vaccine trials.
Prior to 1986, there were 3 routine vaccines totaling 7 injections. The financial liability from these products resulted in companies exiting the market and leaving only one company selling each with the threat they too would cease selling these products. Instead of allowing economic interests to drive innovation of safer products, the 1986 Act gave pharmaceutical companies immunity for vaccine injuries for those products and any childhood vaccine added to CDC’s schedule thereafter. CDC’s maternal and childhood schedules now lists 19 vaccines totaling 84 injections.
Covid-19 vaccines were developed within this same framework. Like other vaccines, companies developing Covid-19 vaccines knew before they were even developed that they would not be liable for injuries they caused – not only because of the 1986 Act, but also because of the additional immunity provided by the PREP Act, which the executive branch guaranteed in the procurement contracts for these products before they were even developed. We are not aware of any other product given the immunity for injuries that has been afforded to vaccine companies.
Economic interests that incentivize a company to assure the safety of its product before release into the market are absent for vaccine products.
Trials for drugs and vaccines are conducted by the pharmaceutical company seeking licensure. For drug products, companies remain liable for injuries the drugs cause after licensure. This provides an incentive to conduct long-term placebo-controlled trials to confirm the safety of drug products before licensure to avoid financial loss after licensure.
In contrast, for vaccine products, this economic incentive was eliminated by the National Childhood Vaccine Injury Act of 1986 (the “1986 Act”) which gave companies immunity for injuries caused by most vaccines. This is why long-term placebo-controlled trials for vaccine products do not make financial sense for companies seeking to maximize profits. In fact, while assuring safety in drug trials is aligned with a company’s economic interest, it is in conflict in vaccine trials.
Prior to 1986, there were 3 routine vaccines totaling 7 injections. The financial liability from these products resulted in companies exiting the market and leaving only one company selling each with the threat they too would cease selling these products. Instead of allowing economic interests to drive innovation of safer products, the 1986 Act gave pharmaceutical companies immunity for vaccine injuries for those products and any childhood vaccine added to CDC’s schedule thereafter. CDC’s maternal and childhood schedules now lists 19 vaccines totaling 84 injections.
Covid-19 vaccines were developed within this same framework. Like other vaccines, companies developing Covid-19 vaccines knew before they were even developed that they would not be liable for injuries they caused – not only because of the 1986 Act, but also because of the additional immunity provided by the PREP Act, which the executive branch guaranteed in the procurement contracts for these products before they were even developed. We are not aware of any other product given the immunity for injuries that has been afforded to vaccine companies.
4.HHS’s Promotion and Defense of Vaccines Conflicts with Regulatory Duties to Identify and Disclose Safety and Efficacy Issues
In recognition that the 1986 Act gutted the economic interest for companies to assure vaccine safety, the 1986 Act made the U.S. Department of Health and Human Services (“HHS”) and its agencies responsible for vaccine safety. The issue is that HHS’s duties to promote and defend vaccines conflict with its safety duties, and the former duties have sublimated the latter such that companies developing Covid-19 vaccines knew they would, even absent a pandemic, face a friendly regulatory environment.
First, duties to promote an industry inherently conflict with duties to identify and address safety issues. This is why, for example, DOT promotes transportation while safety functions are handled by the independent NTSB. Similarly, DOE promotes nuclear power while safety functions are handled by the independent NRC. But with vaccines, these conflicting duties are handled by the same department, HHS. This same conflict exists for Covid-19 vaccines.
Second, HHS is statutorily required to and does vigorously defend against vaccine injury claims. Under the 1986 Act, one can bring a claim for a vaccine injury, but it is against the Secretary of HHS in the Vaccine Injury Compensation Program (“VICP”). This further conflicts HHS, including because any safety issues identified can be used against HHS in the VICP. Vaccines are the only consumer product where the government defends industry against consumers, instead of vice-versa. The same is true for Covid-19 vaccines – the injured are limited to request benefits from HHS in the CICP.
Third, the foregoing conflicts may explain why HHS has failed to perform its basic safety duties pursuant to 42 U.S.C. § 300aa-27, titled Mandate for Safer Childhood Vaccines (the “Mandate”), which underpins vaccine safety in our country. The Mandate has three simple requirements: (i) HHS must submit a biannual report to Congress detailing how it improved vaccine safety in the preceding two years – but it has never filed even one report; (ii) a task force comprised of the heads of CDC, FDA and NIH is to make ongoing recommendations to HHS on how to improve vaccine safety – but that task force was disbanded in 1998; and (iii) a list of HHS’s vaccine safety duties – but its failure to perform the simple foregoing duties belie its performance regarding this far harder duty.
Finally, with regard to FDA and CDC’s independent vaccine advisory committees, VRBPAC and ACIP, a House Report from 2000 found that “[t]he overwhelming majority of members, both voting members and consultants, have substantial ties to the pharmaceutical industry.” An HHS Inspector General report from 2009 found similar issues. Our recent investigation of committee members revealed similar issues.
These structural conflicts in regulating vaccines, whereby regulators view themselves and in fact conduct themselves like partners with pharmaceutical companies when it comes to vaccines (rather than regulators), have deepened since 1986 and are the framework into which Covid-19 vaccines were developed and licensed (as discussed herein) and regulated thereafter (as discussed below).
In recognition that the 1986 Act gutted the economic interest for companies to assure vaccine safety, the 1986 Act made the U.S. Department of Health and Human Services (“HHS”) and its agencies responsible for vaccine safety. The issue is that HHS’s duties to promote and defend vaccines conflict with its safety duties, and the former duties have sublimated the latter such that companies developing Covid-19 vaccines knew they would, even absent a pandemic, face a friendly regulatory environment.
First, duties to promote an industry inherently conflict with duties to identify and address safety issues. This is why, for example, DOT promotes transportation while safety functions are handled by the independent NTSB. Similarly, DOE promotes nuclear power while safety functions are handled by the independent NRC. But with vaccines, these conflicting duties are handled by the same department, HHS. This same conflict exists for Covid-19 vaccines.
Second, HHS is statutorily required to and does vigorously defend against vaccine injury claims. Under the 1986 Act, one can bring a claim for a vaccine injury, but it is against the Secretary of HHS in the Vaccine Injury Compensation Program (“VICP”). This further conflicts HHS, including because any safety issues identified can be used against HHS in the VICP. Vaccines are the only consumer product where the government defends industry against consumers, instead of vice-versa. The same is true for Covid-19 vaccines – the injured are limited to request benefits from HHS in the CICP.
Third, the foregoing conflicts may explain why HHS has failed to perform its basic safety duties pursuant to 42 U.S.C. § 300aa-27, titled Mandate for Safer Childhood Vaccines (the “Mandate”), which underpins vaccine safety in our country. The Mandate has three simple requirements: (i) HHS must submit a biannual report to Congress detailing how it improved vaccine safety in the preceding two years – but it has never filed even one report; (ii) a task force comprised of the heads of CDC, FDA and NIH is to make ongoing recommendations to HHS on how to improve vaccine safety – but that task force was disbanded in 1998; and (iii) a list of HHS’s vaccine safety duties – but its failure to perform the simple foregoing duties belie its performance regarding this far harder duty.
Finally, with regard to FDA and CDC’s independent vaccine advisory committees, VRBPAC and ACIP, a House Report from 2000 found that “[t]he overwhelming majority of members, both voting members and consultants, have substantial ties to the pharmaceutical industry.” An HHS Inspector General report from 2009 found similar issues. Our recent investigation of committee members revealed similar issues.
These structural conflicts in regulating vaccines, whereby regulators view themselves and in fact conduct themselves like partners with pharmaceutical companies when it comes to vaccines (rather than regulators), have deepened since 1986 and are the framework into which Covid-19 vaccines were developed and licensed (as discussed herein) and regulated thereafter (as discussed below).
5.Examples of Structural Conflicts Impacting Covid-19 Vaccine Trials
Clinical trials are supposed to be statistical comparisons of the outcomes of those in the experimental group as compared to those in the placebo group. This avoids, inter alia, the introduction of bias into the trial.
(i)Deaths In Experimental v. Placebo Groups
This statistical comparison approach was used when comparing symptomatic cases in the experimental group (8 cases) and placebo group (162 cases) in Pfizer’s Covid-19 vaccine trial to arrive at a 95% efficacy figure. (It is noted there were 3,410 suspected but unconfirmed cases not included in this analysis, the impact of which remains unknown. ) However, when it came to deaths in the trial, the statistical comparison approach was abandoned and instead each death was judged subjectively.
In July 2021, Pfizer’s published study reported 15 deaths in the vaccinated group and 14 in the placebo group (including 9 cardiovascular deaths in the vaccinated group versus 5 cardiovascular related deaths in the placebo group). In November 2021, FDA’s published report of Pfizer’s trial stated that “there were a total of 38 deaths, 21 in the COMIRNATY [Pfizer’s Covid-19 vaccine] group and 17 in the placebo group. None of the deaths were considered related to vaccination.”
Hence, a statistical comparison was conducted when the data supported the desired conclusion but a subjective assessment when it didn’t. We therefore asked the FDA: “Why are the death data from a randomized controlled trial (‘RCT’) treated like a clinical case-series rather than an RCT when it comes to assessing causality?” FDA responded that it was “unable to respond substantively at this time due to resource constraints and the ongoing pandemic response.”
(ii)Pfizer Fails to Disclose Serious Adverse Events, FDA Takes No Action
The data submitted to FDA is also unreliable as seen from the case of Maddie de Garay. Maddie is now 15 years old and was seriously injured in Pfizer’s Covid-19 clinical trial for 12-15-year-olds, which included only 1,131 children who received the shot. Maddie’s injuries left her wheelchair-bound and reliant upon a feeding tube, yet Pfizer classified her severe injuries as mere “functional abdominal pain” in its emergency use authorization submission to FDA.
On behalf of Maddie, my firm wrote to FDA four times and provided her medical records, and the de Garays submitted their own comment to FDA about this falsity. Neither our firm nor the de Garays received any response until February 26, 2022, 128 days after we first contacted FDA. FDA’s response contained no explanation for the agency’s over 4-month-long delay in responding and, instead, merely suggested that the de Garays file a VAERS report. The de Garays had already done so, which raises serious concern about the claim that “FDA takes all reports of adverse events potentially related to vaccines seriously” as it contends.
We separately commenced a lawsuit on September 3, 2022 against HHS for FDA’s internal communications related to Maddie de Garay. It revealed that on June 24, 2021, in response to inquiries from the public, FDA finally asked Pfizer about Maddie de Garay. On June 30, 2021, Pfizer for the first time disclosed to FDA Maddie’s serious adverse events, including being wheelchair bound and needing a feeding tube. But Pfizer’s report concluded that “the PI [principal investigator] did not feel that the subject’s symptology [sic] was consistent with a vaccine related adverse event.” As reflected in the email chain, FDA appears to simply accept this conclusion.
All serious adverse events in a clinical trial, whether the sponsor considers them related to the product or not, must be reported to FDA. That the Pfizer Covid-19 vaccine causes an injury should not be surprising – injuries from pharmaceutical products occur. What is concerning is that FDA appears unfazed by Pfizer’s failure to adequately disclose this serious injury. FDA should have taken serious issue with this conduct, and its failure to do so reflects the close partnership between FDA and Pfizer. That Pfizer faced no ramifications for failing to accurately and adequately disclose Maddie’s adverse event, in a clinical trial in which just over 1,000 children received the investigational vaccine, leaves open the question of how many other serious injuries were omitted from the data reported by Pfizer to FDA. (Also note that FDA continues to withhold records in its possession concerning Pfizer’s 12–15-year-old trial in which Maddie participated.)
Clinical trials are supposed to be statistical comparisons of the outcomes of those in the experimental group as compared to those in the placebo group. This avoids, inter alia, the introduction of bias into the trial.
(i)Deaths In Experimental v. Placebo Groups
This statistical comparison approach was used when comparing symptomatic cases in the experimental group (8 cases) and placebo group (162 cases) in Pfizer’s Covid-19 vaccine trial to arrive at a 95% efficacy figure. (It is noted there were 3,410 suspected but unconfirmed cases not included in this analysis, the impact of which remains unknown. ) However, when it came to deaths in the trial, the statistical comparison approach was abandoned and instead each death was judged subjectively.
In July 2021, Pfizer’s published study reported 15 deaths in the vaccinated group and 14 in the placebo group (including 9 cardiovascular deaths in the vaccinated group versus 5 cardiovascular related deaths in the placebo group). In November 2021, FDA’s published report of Pfizer’s trial stated that “there were a total of 38 deaths, 21 in the COMIRNATY [Pfizer’s Covid-19 vaccine] group and 17 in the placebo group. None of the deaths were considered related to vaccination.”
Hence, a statistical comparison was conducted when the data supported the desired conclusion but a subjective assessment when it didn’t. We therefore asked the FDA: “Why are the death data from a randomized controlled trial (‘RCT’) treated like a clinical case-series rather than an RCT when it comes to assessing causality?” FDA responded that it was “unable to respond substantively at this time due to resource constraints and the ongoing pandemic response.”
(ii)Pfizer Fails to Disclose Serious Adverse Events, FDA Takes No Action
The data submitted to FDA is also unreliable as seen from the case of Maddie de Garay. Maddie is now 15 years old and was seriously injured in Pfizer’s Covid-19 clinical trial for 12-15-year-olds, which included only 1,131 children who received the shot. Maddie’s injuries left her wheelchair-bound and reliant upon a feeding tube, yet Pfizer classified her severe injuries as mere “functional abdominal pain” in its emergency use authorization submission to FDA.
On behalf of Maddie, my firm wrote to FDA four times and provided her medical records, and the de Garays submitted their own comment to FDA about this falsity. Neither our firm nor the de Garays received any response until February 26, 2022, 128 days after we first contacted FDA. FDA’s response contained no explanation for the agency’s over 4-month-long delay in responding and, instead, merely suggested that the de Garays file a VAERS report. The de Garays had already done so, which raises serious concern about the claim that “FDA takes all reports of adverse events potentially related to vaccines seriously” as it contends.
We separately commenced a lawsuit on September 3, 2022 against HHS for FDA’s internal communications related to Maddie de Garay. It revealed that on June 24, 2021, in response to inquiries from the public, FDA finally asked Pfizer about Maddie de Garay. On June 30, 2021, Pfizer for the first time disclosed to FDA Maddie’s serious adverse events, including being wheelchair bound and needing a feeding tube. But Pfizer’s report concluded that “the PI [principal investigator] did not feel that the subject’s symptology [sic] was consistent with a vaccine related adverse event.” As reflected in the email chain, FDA appears to simply accept this conclusion.
All serious adverse events in a clinical trial, whether the sponsor considers them related to the product or not, must be reported to FDA. That the Pfizer Covid-19 vaccine causes an injury should not be surprising – injuries from pharmaceutical products occur. What is concerning is that FDA appears unfazed by Pfizer’s failure to adequately disclose this serious injury. FDA should have taken serious issue with this conduct, and its failure to do so reflects the close partnership between FDA and Pfizer. That Pfizer faced no ramifications for failing to accurately and adequately disclose Maddie’s adverse event, in a clinical trial in which just over 1,000 children received the investigational vaccine, leaves open the question of how many other serious injuries were omitted from the data reported by Pfizer to FDA. (Also note that FDA continues to withhold records in its possession concerning Pfizer’s 12–15-year-old trial in which Maddie participated.)
6.Preventing Transmission, an Example of Dogma Driving Policy
CDC and FDA should not have been surprised the Covid-19 vaccines did not prevent transmission because even most vaccines mandated for school do not prevent infection and transmission, including inactivated polio vaccine, acellular pertussis vaccine, tetanus vaccine, and meningococcal vaccine. Nor are we aware of a single non-live vaccine for a respiratory infection, like Covid-19 vaccines, that prevents transmission and infection.
As FDA explains, “FDA’s authorization and licensure standards for vaccines do not require demonstration of the prevention of infection or transmission.” FDA nonetheless promoted the belief that the Covid-19 vaccines products could do just that, including in the numerous “Just a Minute” promotional videos released by Dr. Peter Marks in late 2021 and early 2022.
This occurred despite a CDC study, dated August 6, 2021, which found vaccinated individuals had a higher rate of infection and more viral carriage in their nasopharynx than the unvaccinated. With the release of this study, the CDC Director stated on CNN that “what they [Covid-19 vaccines] can’t do anymore is prevent transmission.” Then, on August 24, 2021, a study by the Wisconsin Health Department reviewed swab specimens in 24 counties and found high viral loads in “158 of 232 unvaccinated (68%...) and 156 of 225 fully vaccinated (69%...) symptomatic individuals” and in “7 of 24 unvaccinated (29%...) and 9 of 11 fully vaccinated asymptomatic individuals (82%...).” Our exchange with CDC in mid to late 2021 brought into focus the foregoing.
Nonetheless, the implication these products could prevent infection and transmission persisted, including in a Pfizer report to the FDA on October 26, 2021, stating: “Maximizing the proportion of the population that is vaccinated is critically important to help reduce rates of infection, decrease transmission, prevent the emergence of new variants of concern, and hasten the end of the pandemic.” Despite the lack of clinical evidence to support these claims, FDA permitted Pfizer to continue to make them.
CDC and FDA should not have been surprised the Covid-19 vaccines did not prevent transmission because even most vaccines mandated for school do not prevent infection and transmission, including inactivated polio vaccine, acellular pertussis vaccine, tetanus vaccine, and meningococcal vaccine. Nor are we aware of a single non-live vaccine for a respiratory infection, like Covid-19 vaccines, that prevents transmission and infection.
As FDA explains, “FDA’s authorization and licensure standards for vaccines do not require demonstration of the prevention of infection or transmission.” FDA nonetheless promoted the belief that the Covid-19 vaccines products could do just that, including in the numerous “Just a Minute” promotional videos released by Dr. Peter Marks in late 2021 and early 2022.
This occurred despite a CDC study, dated August 6, 2021, which found vaccinated individuals had a higher rate of infection and more viral carriage in their nasopharynx than the unvaccinated. With the release of this study, the CDC Director stated on CNN that “what they [Covid-19 vaccines] can’t do anymore is prevent transmission.” Then, on August 24, 2021, a study by the Wisconsin Health Department reviewed swab specimens in 24 counties and found high viral loads in “158 of 232 unvaccinated (68%...) and 156 of 225 fully vaccinated (69%...) symptomatic individuals” and in “7 of 24 unvaccinated (29%...) and 9 of 11 fully vaccinated asymptomatic individuals (82%...).” Our exchange with CDC in mid to late 2021 brought into focus the foregoing.
Nonetheless, the implication these products could prevent infection and transmission persisted, including in a Pfizer report to the FDA on October 26, 2021, stating: “Maximizing the proportion of the population that is vaccinated is critically important to help reduce rates of infection, decrease transmission, prevent the emergence of new variants of concern, and hasten the end of the pandemic.” Despite the lack of clinical evidence to support these claims, FDA permitted Pfizer to continue to make them.
7.FDA and CDC Hide Concerning Post-Licensure Safety Data from Public
CDC’s website states that:
"[COVID-19] vaccines are monitored by VAERS and several other vaccine safety monitoring systems as part of the most intensive vaccine safety monitoring effort in U.S. history. This continuous, robust safety monitoring helps keep COVID-19 vaccines safe and helps ensure the benefits of vaccination continue to outweigh any risks."
The other safety monitoring systems are V-safe, CISA, and VSD.
(i)VAERS
To monitor vaccine safety, federal health authorities heavily rely on the Vaccine Adverse Event Reporting System (“VAERS”), a passive vaccine safety surveillance system to which reports of adverse events after vaccination can be submitted. VAERS is co-managed by CDC and FDA.
On December 4, 2020, before the first Covid-19 vaccine was rolled out, CDC released the VAERS Standard Operating Procedures for Covid-19 (“VAERS SOP”), which states in relevant part:
"The analyses for COVID-19 vaccine safety signals will focus on identifying deviations from preliminary safety data, and possibly from other vaccines, using disproportionality analyses and comparisons of reporting rates.
Two main approaches to data mining are Proportional Reporting Ratios (PRRs) and Empirical Bayesian Geometric Means. Both have published literature suggesting criteria for detecting “signals”. PRR will be used at CDC for potential signal detection; Empirical Bayesian data mining will be performed by FDA."
This SOP made clear that CDC planned to conduct safety signal monitoring using Proportional Reporting Ratios (“PRR”) and FDA planned to conduct safety signal monitoring using Empirical Bayesian (“EB”) data mining.
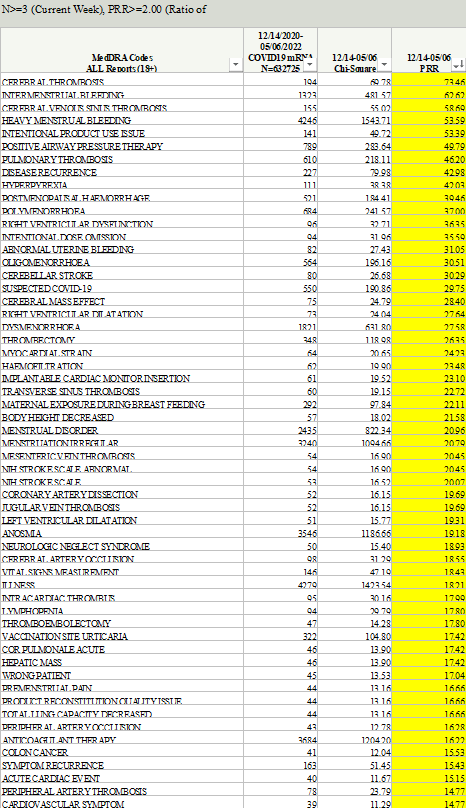
Our firm requested the PRR signal detection data from CDC through FOIA and was denied. In the denial letter, CDC stated that it had not conducted PRR analyses; it instead highlighted the superiority of and historical use of EB data mining, calling it the “gold standard” and the “superior method” with which to detect safety signals. However, on September 2, 2022, then-CDC Director Rochelle Walensky sent a letter to Senator Ron Johnson acknowledging that PRR had in fact been used: “CDC performed PRR analysis between March 25, 2022, through July 31, 2022, to corroborate the results of EB data mining. Notably, results from PRR analysis were generally consistent with EB data mining, revealing no additional unexpected safety signals.” Our firm then sued CDC based on this admission and ultimately received 51 excel files containing PRR data. These files showed that CDC’s own threshold for triggering a signal for adverse events was more than met for numerous serious adverse events, including as seen in the following CDC tables noting that CDC considered, when truth was original, anything above a “2” in the PRR row a safety signal, as provided in the VAERS SOP:
When the CDC was confronted with the above data it sought to hide from the public, it advised Senator Johnson that it was no longer relying upon PRR and instead would only rely upon FDA’s EB data mining; as CDC Director wrote to Senator Johnson:
“CDC and the Food and Drug Administration (FDA) chose to rely on Empirical Bayesian (EB) data mining—a more robust technique used to analyze disproportionate reporting—rather than PRR calculations to mitigate potential false signals. . . . Given the strength of the EB data mining method, CDC and FDA plan to continue relying upon EB data mining moving forward.”
Given that CDC decided to abandon the PRR data and rely instead solely on the EB data, our firm requested the EB data mining results from FDA through FOIA and was denied. Hence, we commenced litigation and the FDA filed a motion requesting that the litigation be stayed for at least 18 months due to the agency being overwhelmed as a result of another court order, issued to our client and litigated by our firm, that ordered FDA to disclose all of the clinical trial documents related to the Pfizer and Moderna Covid-19 vaccines’ licensures. The Court granted the stay for 6 months and then recently granted an additional 6 months. To date, FDA has refused to produce the EB data mining results to the public despite the concerning results shown in the PRR data and despite the fact that it has already located and identified at least 150 records (75 emails and 75 excel files) that are responsive to the request.
(ii)V-safe:
V-safe is CDC’s premier system for tracking the safety of COVID-19 vaccines. It is a smartphone-based program that allows vaccine recipients to “tell CDC about any side effects after getting the COVID-19 vaccine.” Its purpose, as explained by CDC, “is to rapidly characterize the safety profile of COVID-19 vaccines when given outside a clinical trial setting and to detect and evaluate clinically important adverse events and safety issues that might impact policy or regulatory decisions.”
On November 19, 2020, CDC published a protocol for developing v-safe titled “V-safe active surveillance for COVID-19 vaccine safety” which explained that “[t]he purpose of v-safe surveillance is to rapidly characterize the safety profile of COVID-19 vaccines when given outside a clinical trial setting and to detect and evaluate clinically important adverse events and safety issues that might impact policy or regulatory decisions.”
V-safe was launched simultaneously with the EUA of the first COVID-19 vaccine in December 2020. Nine million of the approximate 10 million users who registered for v-safe did so between December 2020 and April 2021. The data submitted by 10 million v-safe users is likely a good reflection of the experience of the larger population of 265 million Americans who received at least one dose of a COVID-19 vaccine.
V-safe collected a limited amount of safety information from its approximately 10 million users using check-the-box options. However, the program also provided a few free-text fields for users to provide additional safety information. CDC received at least 7.8 million free-text entries from v-safe users.
Regarding symptoms collected using check-the-box options, v-safe users are asked to select one or more of 10 listed symptoms that occurred within the first week after vaccination. These symptoms are those that CDC says are normal after vaccination and are actually a sign the vaccine is working by producing an immune response. As CDC explains: “Any side effects from getting the vaccine are normal signs the body is building protection.” The 10 million v-safe users reported over 70 million check-the-box symptoms and this, as expected, did not raise any concerns for CDC as seen from the numerous studies CDC published with this data evidencing these high rates.
The only other check-the-box safety information collected was whether users reported needing medical care, missed school or work, or could not perform normal daily activities. If a user selected that he or she needed medical care, the user was asked to select whether he or she sought telehealth, urgent care, emergency care, or were hospitalized.
Since 2021, CDC has published dozens of studies to support its claim that COVID-19 vaccines are safe. The main data used in these studies is v-safe’s health impact data, with a focus on the rate of people who reported needing medical care after the vaccine. The studies formed the core of CDC’s support for the safety of COVID-19 vaccines, however, they only report the first week of health impact data after injection. This is, at best, highly misleading because CDC is well aware that injuries from COVID-19 vaccines can occur well after the first week.
When CDC finally released the check-the-box data to the public, after over two years of legal demands and a federal lawsuit brought by our firm, the data it hid from the public for over two years showed that 7.7% of v-safe users reported needing medical care after a Covid-19 vaccine (and on average 2 to 3 times per person) and an additional 25% of v-safe users reported missing school or work or being unable to perform normal activities after the injection.
CDC could have made v-safe a rapid and robust safety system by simply including check-the-box options for adverse events of concern (e.g., a check-the-box option for myocarditis or chest pain). In fact, the first version of the V-Safe Protocol, prior to the program’s launch, identified adverse events of special interest (“AESI”) in a chart titled Prespecified Medical Conditions:
Despite CDC itself directly identifying these adverse events as harms of special interest, it did not include check-the-box options for these harms or for common symptoms from these harms.
CDC could have taken advantage of this incredible opportunity – wherein v-safe was already capturing health data from over 10 million users – to easily include these AESIs as check-the-box options for v-safe users. This would have enabled CDC and the scientific community to easily calculate a rate for which v-safe users had myocarditis, or other adverse events that had been prespecified by CDC as potential problems (e.g., strokes, seizures, etc.).
Instead, CDC chose to limit potential reporting of any such adverse events to the free-text fields knowing that, among other issues, fewer people would report issues in a free-text field (versus a check-the-box option) and this free-text data would be more difficult to standardize. In that regard, it does not appear that CDC designed this system with the interests of the public in mind, but rather its own interest to assure control of the data so it can release only data which comports with its a priori policy decision that these products are “safe.”
Nonetheless, a FOIA for the free-text data was submitted and was also heavily litigated initially by another group, and thereafter our firm got involved in that litigation as well and, ultimately, we obtained a Court order that requires CDC to produce the millions of free-text fields on a rolling basis. That production has begun and continues today and through the end of this year.
(iii)CISA
CDC regularly claims that the Clinical Immunization Safety Assessment (“CISA”) is a critical part of the safety monitoring of vaccines. CDC describes CISA as: “a national collaborating network of vaccine safety experts from the CDC’s Immunization Safety Office (ISO), eight medical research centers, and other partners” that was established “to improve the understanding of adverse events following immunization at the individual patient level.” CISA, like the other safety surveillance programs, is also problematic for a few reasons.
For one, as CDC states, “CISA provides consultations for U.S. healthcare providers with complex vaccine safety questions about their patients.” Our firm has heard time and again during the Covid-19 vaccine rollout that many people who suffered adverse events after their vaccination were not believed or being treated by their doctors. Many in the medical field would not acknowledge that the injury could potentially be a vaccine injury and so those people were unable to utilize CISA as it provides consultations only to healthcare providers and not to individual patients.
Moreover, the Principal Investigator of CISA, Dr. Katherine Edwards, was a paid advisor to Pfizer, was compensated by numerous other pharmaceutical companies as a consultant and/or advisor, and also was one of five members of Pfizer Covid-19 vaccine trial’s data safety monitoring board. As explained by bioethicist Arthur Caplan, these boards are “very powerful. They’re key guardians of science and safety and are as important if not more important than the FDA.” Dr. Edwards had a close look at the Pfizer vaccine trial and the ability to stop the trial if there were safety concerns. Following the release of that same product, she was consulting with healthcare providers as to whether or not that same product was the cause of their patients’ serious injuries. This conflict casts serious doubt on the entire CISA program.
(iv)VSD
The Vaccine Safety Datalink is used by CDC and FDA to assess the safety of vaccines. VSD uses electronic health data from participating healthcare organizations and networks throughout the country. The VSD was once maintained at HHS but HHS moved the VSD to a health industry trade association starting in 2001 to avoid having the VSD data subject to FOIA, and to otherwise assure that only the scientists and studies of which it approves utilize the VSD. Thus, when a VSD study is conducted by HHS, in violation of basic scientific standards and process, the underlying raw data is almost never available for inspection by the public and other scientists. So while VSD data is heavily cited and relied upon by the federal health authorities, there is no public access to the data. There are other concerns with VSD as well, such as its lack of ability to assess the long-term impacts of vaccination and its use by the same agency that must defend against claims of vaccine harms, as discussed above.
CDC’s website states that:
"[COVID-19] vaccines are monitored by VAERS and several other vaccine safety monitoring systems as part of the most intensive vaccine safety monitoring effort in U.S. history. This continuous, robust safety monitoring helps keep COVID-19 vaccines safe and helps ensure the benefits of vaccination continue to outweigh any risks."
The other safety monitoring systems are V-safe, CISA, and VSD.
(i)VAERS
To monitor vaccine safety, federal health authorities heavily rely on the Vaccine Adverse Event Reporting System (“VAERS”), a passive vaccine safety surveillance system to which reports of adverse events after vaccination can be submitted. VAERS is co-managed by CDC and FDA.
On December 4, 2020, before the first Covid-19 vaccine was rolled out, CDC released the VAERS Standard Operating Procedures for Covid-19 (“VAERS SOP”), which states in relevant part:
"The analyses for COVID-19 vaccine safety signals will focus on identifying deviations from preliminary safety data, and possibly from other vaccines, using disproportionality analyses and comparisons of reporting rates.
Two main approaches to data mining are Proportional Reporting Ratios (PRRs) and Empirical Bayesian Geometric Means. Both have published literature suggesting criteria for detecting “signals”. PRR will be used at CDC for potential signal detection; Empirical Bayesian data mining will be performed by FDA."
This SOP made clear that CDC planned to conduct safety signal monitoring using Proportional Reporting Ratios (“PRR”) and FDA planned to conduct safety signal monitoring using Empirical Bayesian (“EB”) data mining.
Our firm requested the PRR signal detection data from CDC through FOIA and was denied. In the denial letter, CDC stated that it had not conducted PRR analyses; it instead highlighted the superiority of and historical use of EB data mining, calling it the “gold standard” and the “superior method” with which to detect safety signals. However, on September 2, 2022, then-CDC Director Rochelle Walensky sent a letter to Senator Ron Johnson acknowledging that PRR had in fact been used: “CDC performed PRR analysis between March 25, 2022, through July 31, 2022, to corroborate the results of EB data mining. Notably, results from PRR analysis were generally consistent with EB data mining, revealing no additional unexpected safety signals.” Our firm then sued CDC based on this admission and ultimately received 51 excel files containing PRR data. These files showed that CDC’s own threshold for triggering a signal for adverse events was more than met for numerous serious adverse events, including as seen in the following CDC tables noting that CDC considered, when truth was original, anything above a “2” in the PRR row a safety signal, as provided in the VAERS SOP:
When the CDC was confronted with the above data it sought to hide from the public, it advised Senator Johnson that it was no longer relying upon PRR and instead would only rely upon FDA’s EB data mining; as CDC Director wrote to Senator Johnson:
“CDC and the Food and Drug Administration (FDA) chose to rely on Empirical Bayesian (EB) data mining—a more robust technique used to analyze disproportionate reporting—rather than PRR calculations to mitigate potential false signals. . . . Given the strength of the EB data mining method, CDC and FDA plan to continue relying upon EB data mining moving forward.”
Given that CDC decided to abandon the PRR data and rely instead solely on the EB data, our firm requested the EB data mining results from FDA through FOIA and was denied. Hence, we commenced litigation and the FDA filed a motion requesting that the litigation be stayed for at least 18 months due to the agency being overwhelmed as a result of another court order, issued to our client and litigated by our firm, that ordered FDA to disclose all of the clinical trial documents related to the Pfizer and Moderna Covid-19 vaccines’ licensures. The Court granted the stay for 6 months and then recently granted an additional 6 months. To date, FDA has refused to produce the EB data mining results to the public despite the concerning results shown in the PRR data and despite the fact that it has already located and identified at least 150 records (75 emails and 75 excel files) that are responsive to the request.
(ii)V-safe:
V-safe is CDC’s premier system for tracking the safety of COVID-19 vaccines. It is a smartphone-based program that allows vaccine recipients to “tell CDC about any side effects after getting the COVID-19 vaccine.” Its purpose, as explained by CDC, “is to rapidly characterize the safety profile of COVID-19 vaccines when given outside a clinical trial setting and to detect and evaluate clinically important adverse events and safety issues that might impact policy or regulatory decisions.”
On November 19, 2020, CDC published a protocol for developing v-safe titled “V-safe active surveillance for COVID-19 vaccine safety” which explained that “[t]he purpose of v-safe surveillance is to rapidly characterize the safety profile of COVID-19 vaccines when given outside a clinical trial setting and to detect and evaluate clinically important adverse events and safety issues that might impact policy or regulatory decisions.”
V-safe was launched simultaneously with the EUA of the first COVID-19 vaccine in December 2020. Nine million of the approximate 10 million users who registered for v-safe did so between December 2020 and April 2021. The data submitted by 10 million v-safe users is likely a good reflection of the experience of the larger population of 265 million Americans who received at least one dose of a COVID-19 vaccine.
V-safe collected a limited amount of safety information from its approximately 10 million users using check-the-box options. However, the program also provided a few free-text fields for users to provide additional safety information. CDC received at least 7.8 million free-text entries from v-safe users.
Regarding symptoms collected using check-the-box options, v-safe users are asked to select one or more of 10 listed symptoms that occurred within the first week after vaccination. These symptoms are those that CDC says are normal after vaccination and are actually a sign the vaccine is working by producing an immune response. As CDC explains: “Any side effects from getting the vaccine are normal signs the body is building protection.” The 10 million v-safe users reported over 70 million check-the-box symptoms and this, as expected, did not raise any concerns for CDC as seen from the numerous studies CDC published with this data evidencing these high rates.
The only other check-the-box safety information collected was whether users reported needing medical care, missed school or work, or could not perform normal daily activities. If a user selected that he or she needed medical care, the user was asked to select whether he or she sought telehealth, urgent care, emergency care, or were hospitalized.
Since 2021, CDC has published dozens of studies to support its claim that COVID-19 vaccines are safe. The main data used in these studies is v-safe’s health impact data, with a focus on the rate of people who reported needing medical care after the vaccine. The studies formed the core of CDC’s support for the safety of COVID-19 vaccines, however, they only report the first week of health impact data after injection. This is, at best, highly misleading because CDC is well aware that injuries from COVID-19 vaccines can occur well after the first week.
When CDC finally released the check-the-box data to the public, after over two years of legal demands and a federal lawsuit brought by our firm, the data it hid from the public for over two years showed that 7.7% of v-safe users reported needing medical care after a Covid-19 vaccine (and on average 2 to 3 times per person) and an additional 25% of v-safe users reported missing school or work or being unable to perform normal activities after the injection.
CDC could have made v-safe a rapid and robust safety system by simply including check-the-box options for adverse events of concern (e.g., a check-the-box option for myocarditis or chest pain). In fact, the first version of the V-Safe Protocol, prior to the program’s launch, identified adverse events of special interest (“AESI”) in a chart titled Prespecified Medical Conditions:
Despite CDC itself directly identifying these adverse events as harms of special interest, it did not include check-the-box options for these harms or for common symptoms from these harms.
CDC could have taken advantage of this incredible opportunity – wherein v-safe was already capturing health data from over 10 million users – to easily include these AESIs as check-the-box options for v-safe users. This would have enabled CDC and the scientific community to easily calculate a rate for which v-safe users had myocarditis, or other adverse events that had been prespecified by CDC as potential problems (e.g., strokes, seizures, etc.).
Instead, CDC chose to limit potential reporting of any such adverse events to the free-text fields knowing that, among other issues, fewer people would report issues in a free-text field (versus a check-the-box option) and this free-text data would be more difficult to standardize. In that regard, it does not appear that CDC designed this system with the interests of the public in mind, but rather its own interest to assure control of the data so it can release only data which comports with its a priori policy decision that these products are “safe.”
Nonetheless, a FOIA for the free-text data was submitted and was also heavily litigated initially by another group, and thereafter our firm got involved in that litigation as well and, ultimately, we obtained a Court order that requires CDC to produce the millions of free-text fields on a rolling basis. That production has begun and continues today and through the end of this year.
(iii)CISA
CDC regularly claims that the Clinical Immunization Safety Assessment (“CISA”) is a critical part of the safety monitoring of vaccines. CDC describes CISA as: “a national collaborating network of vaccine safety experts from the CDC’s Immunization Safety Office (ISO), eight medical research centers, and other partners” that was established “to improve the understanding of adverse events following immunization at the individual patient level.” CISA, like the other safety surveillance programs, is also problematic for a few reasons.
For one, as CDC states, “CISA provides consultations for U.S. healthcare providers with complex vaccine safety questions about their patients.” Our firm has heard time and again during the Covid-19 vaccine rollout that many people who suffered adverse events after their vaccination were not believed or being treated by their doctors. Many in the medical field would not acknowledge that the injury could potentially be a vaccine injury and so those people were unable to utilize CISA as it provides consultations only to healthcare providers and not to individual patients.
Moreover, the Principal Investigator of CISA, Dr. Katherine Edwards, was a paid advisor to Pfizer, was compensated by numerous other pharmaceutical companies as a consultant and/or advisor, and also was one of five members of Pfizer Covid-19 vaccine trial’s data safety monitoring board. As explained by bioethicist Arthur Caplan, these boards are “very powerful. They’re key guardians of science and safety and are as important if not more important than the FDA.” Dr. Edwards had a close look at the Pfizer vaccine trial and the ability to stop the trial if there were safety concerns. Following the release of that same product, she was consulting with healthcare providers as to whether or not that same product was the cause of their patients’ serious injuries. This conflict casts serious doubt on the entire CISA program.
(iv)VSD
The Vaccine Safety Datalink is used by CDC and FDA to assess the safety of vaccines. VSD uses electronic health data from participating healthcare organizations and networks throughout the country. The VSD was once maintained at HHS but HHS moved the VSD to a health industry trade association starting in 2001 to avoid having the VSD data subject to FOIA, and to otherwise assure that only the scientists and studies of which it approves utilize the VSD. Thus, when a VSD study is conducted by HHS, in violation of basic scientific standards and process, the underlying raw data is almost never available for inspection by the public and other scientists. So while VSD data is heavily cited and relied upon by the federal health authorities, there is no public access to the data. There are other concerns with VSD as well, such as its lack of ability to assess the long-term impacts of vaccination and its use by the same agency that must defend against claims of vaccine harms, as discussed above.

Full version with footnotes available at:
Link to video of full hearing: judiciary.house.gov/sites/evo-subs…
judiciary.house.gov/committee-acti…
Link to video of full hearing: judiciary.house.gov/sites/evo-subs…
judiciary.house.gov/committee-acti…
• • •
Missing some Tweet in this thread? You can try to
force a refresh






