Today parents can, for the first time, access polygenic embryo testing for IQ and many diseases through a routine test (PGT-A) done worldwide and in 60% of US IVF cycles.
Read on to learn how Herasight’s ImputePGTA algorithm is enabling polygenic testing in IVF to go global. 🧵
Read on to learn how Herasight’s ImputePGTA algorithm is enabling polygenic testing in IVF to go global. 🧵

Done IVF or considering IVF? Sign up here:
Herasight has already served people from 11 countries across Europe, North America, and Asia.
Now we’re offering a 90% discount to the first qualified customer from each country in the world (T&Cs apply). herasight.com/get-early-acce…
Herasight has already served people from 11 countries across Europe, North America, and Asia.
Now we’re offering a 90% discount to the first qualified customer from each country in the world (T&Cs apply). herasight.com/get-early-acce…

PGT-A tests for chromosomal abnormalities and is routine in the US, with rates increasing in recent years, and is common globally.
Furthermore, almost 1 million frozen embryos in the US already have PGT-A data, which can now be used for polygenic testing through Herasight.
Furthermore, almost 1 million frozen embryos in the US already have PGT-A data, which can now be used for polygenic testing through Herasight.

Previously, couples wanting to access polygenic embryo testing (PGT-P) could only do so by undergoing specialized wet-lab procedures provided by two US companies.
Herasight’s technical innovation is breaking this duopoly and democratizing access worldwide.
Herasight’s technical innovation is breaking this duopoly and democratizing access worldwide.
In many jurisdictions you have a legal right to your PGT-A data under regulations such as HIPAA in the USA and GDPR in the EU.
Herasight has a lot of experience dealing with these bureaucratic processes and gatekeepers. Contact us () for help. herasight.com/get-early-acce…
Herasight has a lot of experience dealing with these bureaucratic processes and gatekeepers. Contact us () for help. herasight.com/get-early-acce…

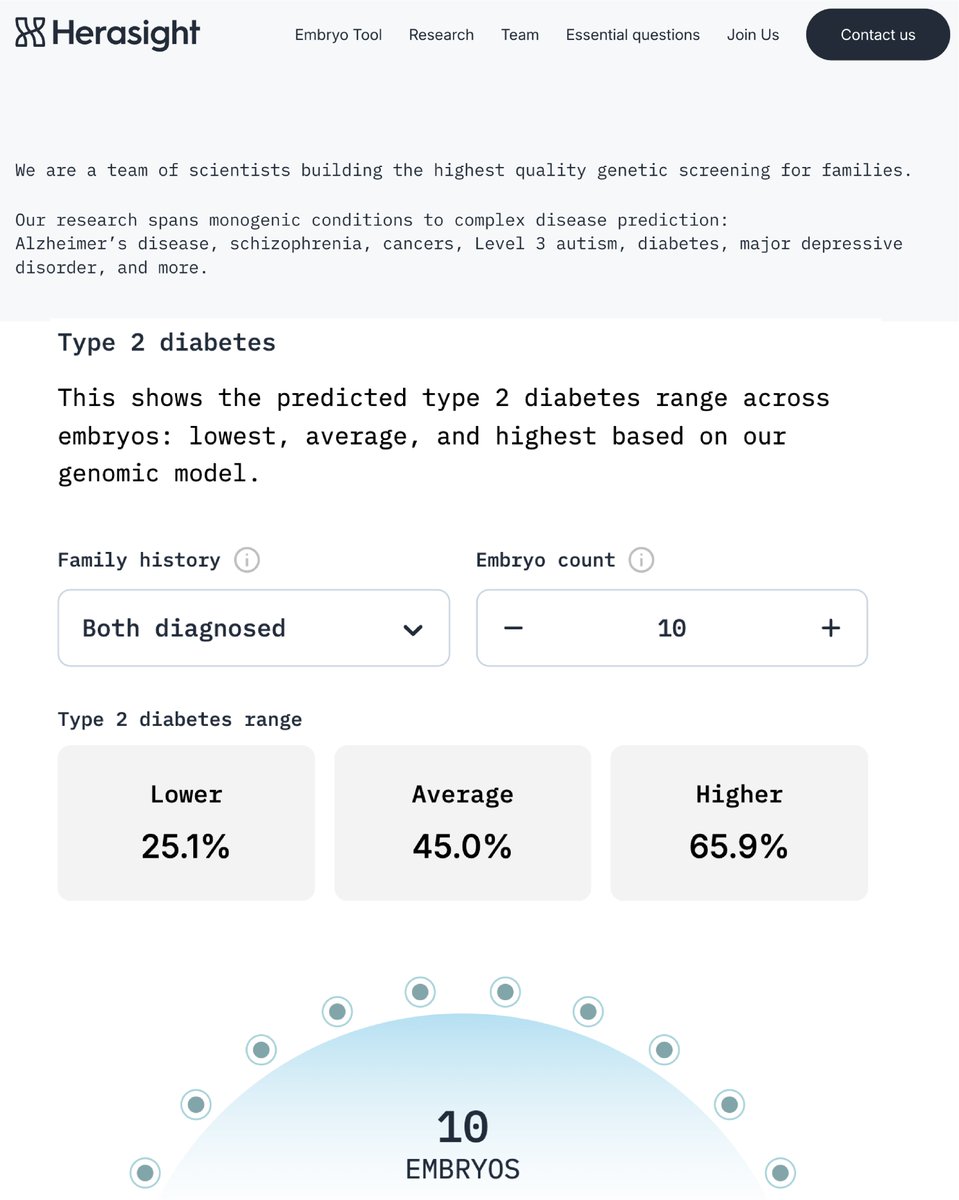
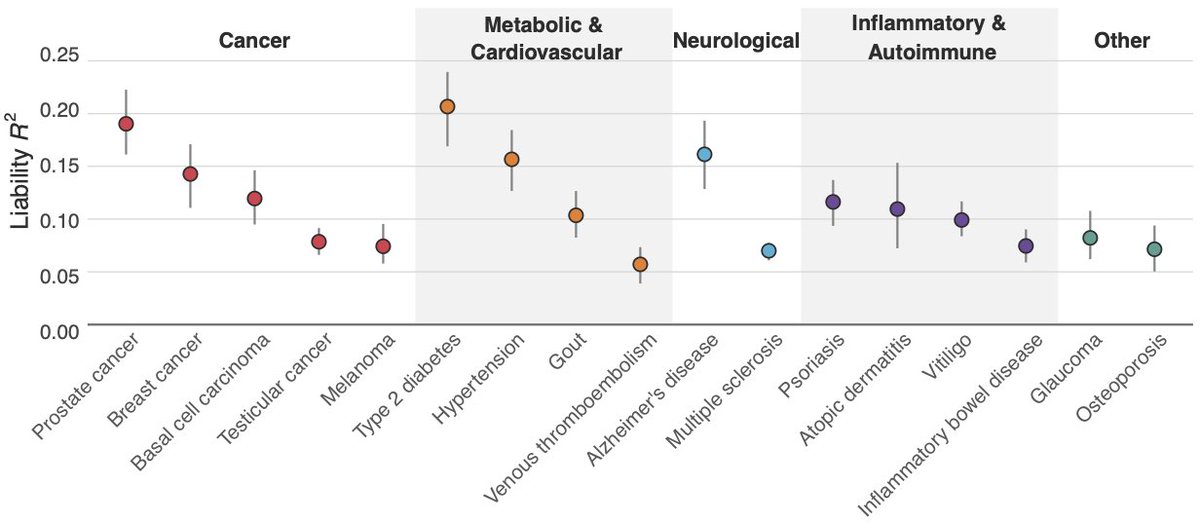
Couples with PGT-A data can now access Herasight’s polygenic predictors for IQ and many diseases, including the most powerful predictors for Alzheimer’s, schizophrenia, and cancers.
Selection on our IQ predictor, CogPGT, can boost offspring IQ by up to 9 points: see
Selection on our IQ predictor, CogPGT, can boost offspring IQ by up to 9 points: see
https://x.com/SponceyM/status/1980660198441447568

For example, someone in Mongolia reached out to Herasight after hearing about us on Mongolian radio.
By sending us their PGT-A data along with parental saliva samples, we can provide them with advanced genetic testing of embryos previously only available in the US.
By sending us their PGT-A data along with parental saliva samples, we can provide them with advanced genetic testing of embryos previously only available in the US.

Read this thread and our white paper () for all the technical details.
Pre-implantation genetic testing for aneuploidy (PGT-A) uses ultra-low-pass embryo sequencing (or, more rarely, genotyping array data on embryos) to detect chromosomal abnormalities. tinyurl.com/wszncyzc
Pre-implantation genetic testing for aneuploidy (PGT-A) uses ultra-low-pass embryo sequencing (or, more rarely, genotyping array data on embryos) to detect chromosomal abnormalities. tinyurl.com/wszncyzc

PGT-A data is orders of magnitude sparser than that used by standard methods for genotype imputation.
However, in IVF we have access to the ultimate reference genomes: the parents.
Each embryo’s genome is a mosaic of its parents' chromosomes (plus de novo mutations).
However, in IVF we have access to the ultimate reference genomes: the parents.
Each embryo’s genome is a mosaic of its parents' chromosomes (plus de novo mutations).

Each parent carries two copies of each chromosome (i.e. two haplotypes).
We can reconstruct the embryo’s genome by using PGT-A data to infer which parental haplotypes the embryo inherits at each position in the genome.
We can reconstruct the embryo’s genome by using PGT-A data to infer which parental haplotypes the embryo inherits at each position in the genome.

However, we don’t know the true parental haplotypes.
Instead, haplotypes are estimated using statistical phasing. But this is imperfect and results in estimated haplotypes that are mosaics of the true haplotypes, with ‘switch errors’ being the points where they flip over.
Instead, haplotypes are estimated using statistical phasing. But this is imperfect and results in estimated haplotypes that are mosaics of the true haplotypes, with ‘switch errors’ being the points where they flip over.

Switch-errors make the technical problem much more challenging than if we knew the true parental haplotypes since true recombinations, where the true parental haplotype the offspring inherits changes, are rare, but switch-errors can occur very frequently.
We developed a combined wet-lab and computational pipeline to produce high-quality estimated parental haplotypes.
Using a novel algorithm developed by @jeremyli__, we combine statistical phasing from short-read sequence data with read-backed phasing from Oxford Nanopore long-reads.
Importantly, although long-read phasing is not necessary for polygenic embryo scoring, it allows us to phase rare variants not present in a reference panel, which enables us to infer rare variants carried by embryos, not just common variants.
Using a novel algorithm developed by @jeremyli__, we combine statistical phasing from short-read sequence data with read-backed phasing from Oxford Nanopore long-reads.
Importantly, although long-read phasing is not necessary for polygenic embryo scoring, it allows us to phase rare variants not present in a reference panel, which enables us to infer rare variants carried by embryos, not just common variants.

Given the estimated parental haplotypes, offspring inheritance patterns are inferred using our innovative Hidden Markov Model based algorithm, ImputePGTA.
ImputePGTA gives posterior distributions over offspring inheritance vectors, allowing us to properly account for uncertainty.
ImputePGTA gives posterior distributions over offspring inheritance vectors, allowing us to properly account for uncertainty.

In simulated data we found that, provided parental phasing quality was good, ImputePGTA achieved highly accurate embryo genotypes (correlation ~0.95) and polygenic scores (error ~0.2SDs) for typical PGT-A coverage (0.004x).

We analysed the theoretical loss of selection efficacy from uncertainty and performed simulations using 17 Herasight disease PGSs, finding a ~5% reduction in efficacy when using PGT-A data w/ long reads on parents, ~10% w/out long reads, in line with theoretical results.
This analysis, based on a real family (the Platinum Pedigree), also shows that the efficacy of polygenic embryo selection can differ substantially from the population average for each family and differ within-family across different diseases.
For example, a parent in this family is heterozygous for an APOE risk allele, which leads to highly increased risk of Alzheimer’s disease. Embryo selection for such families is much more effective than would be predicted from theoretical and simulation results that make simplifying assumptions and compute expected results at the population level.
This analysis, based on a real family (the Platinum Pedigree), also shows that the efficacy of polygenic embryo selection can differ substantially from the population average for each family and differ within-family across different diseases.
For example, a parent in this family is heterozygous for an APOE risk allele, which leads to highly increased risk of Alzheimer’s disease. Embryo selection for such families is much more effective than would be predicted from theoretical and simulation results that make simplifying assumptions and compute expected results at the population level.

We tested our approach using four families that underwent IVF with PGT-A testing, followed by at least one successful pregnancy.
We sought to compare our reconstructed embryo genomes to the ‘true’ genomes of the born child from high quality post-birth sequencing.
We sought to compare our reconstructed embryo genomes to the ‘true’ genomes of the born child from high quality post-birth sequencing.

This figure from FAM1 shows how the quality of parental phasing affects the inference of offspring inheritance vectors (which parental haplotype they inherit from). Long reads improve on short reads, and if we also have grandparents, we obtain ~true parental haplotypes.

This shows posterior distributions for embryo PGSs from real PGTA data vs true embryo PGSs (at 0). The true PGS generally overlapped regions of high posterior density, with a mean-absolute-error of 0.16SDs (0.27SDs short reads only).
Importantly, we propagate uncertainty in embryo PGS values into our trait and disease prediction models.
Importantly, we propagate uncertainty in embryo PGS values into our trait and disease prediction models.

Two families had array based PGTA data, a less common and less technically challenging scenario.
ImputePGTA applied to array data gives highly accurate results in all scenarios, more accurate than imputation from reference, the standard approach in academic research.
ImputePGTA applied to array data gives highly accurate results in all scenarios, more accurate than imputation from reference, the standard approach in academic research.

We already have a more advanced and innovative algorithm working for ImputePGTA v2 that is giving even stronger results, especially for cases where parental phasing is poor. We plan to update our white paper soon with this and data from more real PGT-A families.
Thanks to @jeremyli__, @_twolfram, and everyone @herasight who made this project possible, as well as @ShaiCarmi for comments on the white paper. This project is the reason I got involved with @herasight and it’s great to see it coming to fruition.
• • •
Missing some Tweet in this thread? You can try to
force a refresh