
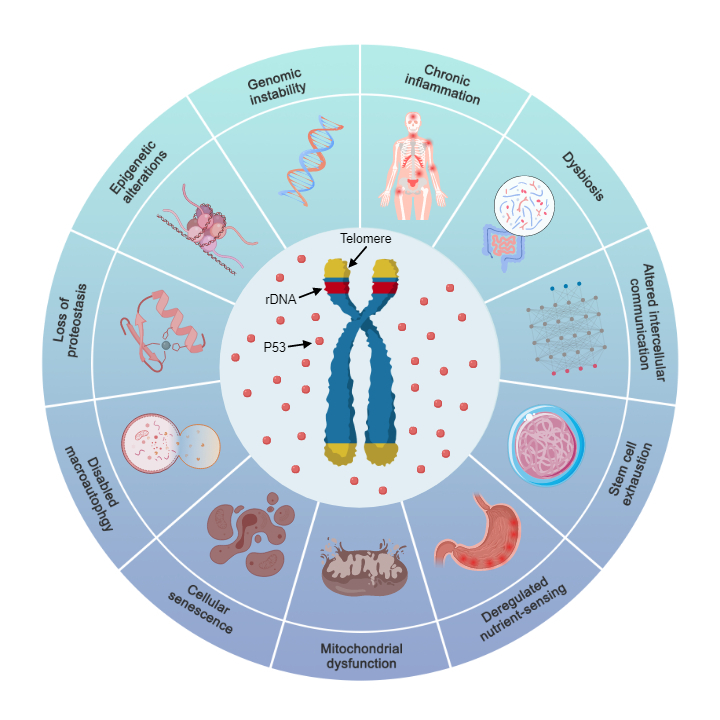
Mitochondrial dysfunction is merely a downstream consequence of telomeres and rDNA driving cellular senescence through the p53 pathway, rather than the root cause of aging.
During cellular senescence, the upregulation of p53 levels not only increases the expression of certain genes and decreases that of others[58], but also causes a continuous decline in the synthesis rates of total proteins and ATP[120−123]. This is because p53 binds to the promoter of the histone deacetylase gene HDAC2 to promote its transcription[124]. HDAC2 mediates histone deacetylation, which tightens the binding between histones and DNA, impairs gene transcription, and consequently reduces the synthesis rate of total proteins. Moreover, more than 80% of the proteins involved in mitochondrial ATP synthesis are encoded by nuclear genes[125]. Accordingly, p53 upregulation leads to a concurrent reduction in the synthesis rates of both total proteins and ATP. The sequential reaction process is as follows:
Telomere and/or rDNA array shortening → p53 upregulation → differential gene expression (partial gene upregulation and downregulation) and HDAC2 upregulation → decreased synthesis rates of total proteins and ATP → decline and alteration of cellular functions.
In conclusion, cellular senescence is not driven by the mutation and accumulation of mitochondrial DNA (mtDNA), and the mitochondrial dysfunction observed in senescent cells does not stem from intrinsic defects of mitochondria themselves.
Reference:
[58] Ergen AV, Goodell MA. Mechanisms of hematopoietic stem cell aging. Exp Gerontol. 2010 Apr;45(4):286-90. doi: 10.1016/j.exger.2009.12.010.
[120] Cook JR, Buetow DE. Decreased protein synthesis by polysomes, tRNA and aminoacyl-tRNA synthetases isolated from senescent rat liver. Mech Ageing Dev. 1981 Sep;17(1):41-52. doi: 10.1016/0047-6374(81)90127-5.
[121] Llewellyn J, Mallikarjun V, Appleton E, et al. Loss of regulation of protein synthesis and turnover underpins an attenuated stress response in senescent human mesenchymal stem cells. Proc Natl Acad Sci U S A. 2023 Apr 4;120(14):e2210745120. doi: 10.1073/pnas.2210745120.
[122] Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005 Apr 12;102(15):5618-23. doi: 10.1073/pnas.0501559102.
[123] Kim JY, Atanassov I, Dethloff F, et al. Time-resolved proteomic analyses of senescence highlight metabolic rewiring of mitochondria. Life Sci Alliance. 2023 Jun 15;6(9):e202302127. doi: 10.26508/lsa.202302127.
[124] Li XL, Zhou J, Chen ZR, et al. P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World J Gastroenterol. 2015 Jan 7;21(1):84-93. doi: 10.3748/wjg.v21.i1.84.
[125] Koopman WJ, Distelmaier F, Smeitink JA, et al. OXPHOS mutations and neurodegeneration. EMBO J. 2013 Jan 9;32(1):9-29. doi: 10.1038/emboj.2012.300.
During cellular senescence, the upregulation of p53 levels not only increases the expression of certain genes and decreases that of others[58], but also causes a continuous decline in the synthesis rates of total proteins and ATP[120−123]. This is because p53 binds to the promoter of the histone deacetylase gene HDAC2 to promote its transcription[124]. HDAC2 mediates histone deacetylation, which tightens the binding between histones and DNA, impairs gene transcription, and consequently reduces the synthesis rate of total proteins. Moreover, more than 80% of the proteins involved in mitochondrial ATP synthesis are encoded by nuclear genes[125]. Accordingly, p53 upregulation leads to a concurrent reduction in the synthesis rates of both total proteins and ATP. The sequential reaction process is as follows:
Telomere and/or rDNA array shortening → p53 upregulation → differential gene expression (partial gene upregulation and downregulation) and HDAC2 upregulation → decreased synthesis rates of total proteins and ATP → decline and alteration of cellular functions.
In conclusion, cellular senescence is not driven by the mutation and accumulation of mitochondrial DNA (mtDNA), and the mitochondrial dysfunction observed in senescent cells does not stem from intrinsic defects of mitochondria themselves.
Reference:
[58] Ergen AV, Goodell MA. Mechanisms of hematopoietic stem cell aging. Exp Gerontol. 2010 Apr;45(4):286-90. doi: 10.1016/j.exger.2009.12.010.
[120] Cook JR, Buetow DE. Decreased protein synthesis by polysomes, tRNA and aminoacyl-tRNA synthetases isolated from senescent rat liver. Mech Ageing Dev. 1981 Sep;17(1):41-52. doi: 10.1016/0047-6374(81)90127-5.
[121] Llewellyn J, Mallikarjun V, Appleton E, et al. Loss of regulation of protein synthesis and turnover underpins an attenuated stress response in senescent human mesenchymal stem cells. Proc Natl Acad Sci U S A. 2023 Apr 4;120(14):e2210745120. doi: 10.1073/pnas.2210745120.
[122] Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005 Apr 12;102(15):5618-23. doi: 10.1073/pnas.0501559102.
[123] Kim JY, Atanassov I, Dethloff F, et al. Time-resolved proteomic analyses of senescence highlight metabolic rewiring of mitochondria. Life Sci Alliance. 2023 Jun 15;6(9):e202302127. doi: 10.26508/lsa.202302127.
[124] Li XL, Zhou J, Chen ZR, et al. P53 mutations in colorectal cancer - molecular pathogenesis and pharmacological reactivation. World J Gastroenterol. 2015 Jan 7;21(1):84-93. doi: 10.3748/wjg.v21.i1.84.
[125] Koopman WJ, Distelmaier F, Smeitink JA, et al. OXPHOS mutations and neurodegeneration. EMBO J. 2013 Jan 9;32(1):9-29. doi: 10.1038/emboj.2012.300.
https://x.com/BiluHuang/status/2048598513752105337

4.3The Mitochondrial Theory of Aging
In 1980, Miquel et al. [85] proposed the mitochondrial theory of cellular aging, which posits that ROS generated by mitochondria can cause oxidative damage to mtDNA, lipids, and proteins within the cell. This damage leads to cellular, tissue, and organ dysfunction, ultimately accelerating the aging process.
However, proteins damaged by ROS can be degraded and renewed, and mutated mtDNA can also be selectively cleared [86]. On April 1, 2002, I published an article titled “Can We Live Forever?” in Science and Technology Daily, in which I wrote: “Abnormal or dysfunctional mitochondria will be recognized and engulfed by lysosomes” [87]. The selective engulfing of mutated mitochondria by lysosomes is called “mitophagy,” a term proposed by Lemasters in 2005 [88]. The reason why senescent cells accumulate mutated mtDNA is that during the aging process, the genetic program intentionally shuts down this solution.
In 1958, Yoshida discovered [89] that within 8 hours, the chloroplasts in the nucleated protoplasts of Elodea densa underwent a process of aging and structural disintegration, while the chloroplasts in the enucleated protoplasts remained green and continued to accumulate starch. In 1975, Wright and Hayflick transplanted young nuclei into enucleated aged cytoplasm, and the cells regained their youth and continued to divide according to the remaining number of divisions of the young nuclei. Conversely, when aged nuclei were transplanted into enucleated young cytoplasm, the cells exhibited an aging phenotype and the number of cell divisions was greatly reduced. These studies indicate that the site determining cellular aging is not the mitochondria, but the cell nucleus. In other words, the accumulation of mtDNA mutations cannot lead to cellular aging. Moreover, increasing telomere length can significantly elevate the number of cell divisions. When mice were treated with AAV expressing mouse TERT at the ages of 1 and 2 years old, their average lifespan was extended by 24% and 13%, respectively [90].
However, there is no evidence to suggest that promoting mitochondrial function can also increase the number of cell divisions and significantly extend the lifespan of mice.
Whether mtDNA mutations cause animal aging or are merely correlated with it is a matter of intense debate. The following are several pieces of evidence that do not support the mitochondrial theory of cellular aging:
The mtDNA mutations in the oocytes of rhesus monkeys and humans do not accumulate with age [91]; Vermulst et al. [92] found that mice models with increased mtDNA mutations showed no signs of accelerated aging and did not have shortened lifespans; Heterozygous mutation of mouse mitochondrial superoxide dismutase (SOD2) led to increased oxidative damage and mtDNA mutations, but did not shorten the animals' lifespans [93]; The main characteristic of mitochondrial dysfunction is a decrease in ATP production. However, when doxycycline was used to inhibit ATP production by the mitochondria in Caenorhabditis elegans, the lifespan of the treated group was extended by 72.8%. In contrast, treatment with vitamin C, which led to a 2.5 - fold increase in ATP production, resulted in an approximately 18% increase in LF content within the worms' bodies after 8 days of treatment, and accelerated aging [49].
Cells and organisms have various solutions for clearing mutated mtDNA: During the reprogramming process, mtDNA mutations in heteroplasmic cells show a bimodal distribution, and after four divisions, they either quickly lose the mutations or acquire more. iPSCs with high - mutation heteroplasmy grow slowly, and their EpiAge increases. This suggests that in tissues, cells with high - mutation mtDNA cannot compete with healthy cells and will be eliminated [94]; Drosophila can selectively clear mitochondria containing mutated mtDNA in muscles through mitophagy [95]; Lysosomes can degrade damaged mitochondria, and when lysosome function is impaired or overwhelmed, fibroblasts and cardiomyocytes will excrete damaged mitochondria through exosomes [96]; Damaged mitochondria in neurons are selectively expelled outside the cell and then taken up and degraded by astrocytes [97]; During the differentiation process, stem cells allocate more defective mitochondria to the progenitor cells of daughter cells and more normal mitochondria to the stem cells of daughter cells [98].
There are many other pieces of evidence that do not support the mitochondrial theory of cellular aging, which are not listed one by one in this paper. In conclusion, the mitochondrial theory of aging is wrong.
In 1980, Miquel et al. [85] proposed the mitochondrial theory of cellular aging, which posits that ROS generated by mitochondria can cause oxidative damage to mtDNA, lipids, and proteins within the cell. This damage leads to cellular, tissue, and organ dysfunction, ultimately accelerating the aging process.
However, proteins damaged by ROS can be degraded and renewed, and mutated mtDNA can also be selectively cleared [86]. On April 1, 2002, I published an article titled “Can We Live Forever?” in Science and Technology Daily, in which I wrote: “Abnormal or dysfunctional mitochondria will be recognized and engulfed by lysosomes” [87]. The selective engulfing of mutated mitochondria by lysosomes is called “mitophagy,” a term proposed by Lemasters in 2005 [88]. The reason why senescent cells accumulate mutated mtDNA is that during the aging process, the genetic program intentionally shuts down this solution.
In 1958, Yoshida discovered [89] that within 8 hours, the chloroplasts in the nucleated protoplasts of Elodea densa underwent a process of aging and structural disintegration, while the chloroplasts in the enucleated protoplasts remained green and continued to accumulate starch. In 1975, Wright and Hayflick transplanted young nuclei into enucleated aged cytoplasm, and the cells regained their youth and continued to divide according to the remaining number of divisions of the young nuclei. Conversely, when aged nuclei were transplanted into enucleated young cytoplasm, the cells exhibited an aging phenotype and the number of cell divisions was greatly reduced. These studies indicate that the site determining cellular aging is not the mitochondria, but the cell nucleus. In other words, the accumulation of mtDNA mutations cannot lead to cellular aging. Moreover, increasing telomere length can significantly elevate the number of cell divisions. When mice were treated with AAV expressing mouse TERT at the ages of 1 and 2 years old, their average lifespan was extended by 24% and 13%, respectively [90].
However, there is no evidence to suggest that promoting mitochondrial function can also increase the number of cell divisions and significantly extend the lifespan of mice.
Whether mtDNA mutations cause animal aging or are merely correlated with it is a matter of intense debate. The following are several pieces of evidence that do not support the mitochondrial theory of cellular aging:
The mtDNA mutations in the oocytes of rhesus monkeys and humans do not accumulate with age [91]; Vermulst et al. [92] found that mice models with increased mtDNA mutations showed no signs of accelerated aging and did not have shortened lifespans; Heterozygous mutation of mouse mitochondrial superoxide dismutase (SOD2) led to increased oxidative damage and mtDNA mutations, but did not shorten the animals' lifespans [93]; The main characteristic of mitochondrial dysfunction is a decrease in ATP production. However, when doxycycline was used to inhibit ATP production by the mitochondria in Caenorhabditis elegans, the lifespan of the treated group was extended by 72.8%. In contrast, treatment with vitamin C, which led to a 2.5 - fold increase in ATP production, resulted in an approximately 18% increase in LF content within the worms' bodies after 8 days of treatment, and accelerated aging [49].
Cells and organisms have various solutions for clearing mutated mtDNA: During the reprogramming process, mtDNA mutations in heteroplasmic cells show a bimodal distribution, and after four divisions, they either quickly lose the mutations or acquire more. iPSCs with high - mutation heteroplasmy grow slowly, and their EpiAge increases. This suggests that in tissues, cells with high - mutation mtDNA cannot compete with healthy cells and will be eliminated [94]; Drosophila can selectively clear mitochondria containing mutated mtDNA in muscles through mitophagy [95]; Lysosomes can degrade damaged mitochondria, and when lysosome function is impaired or overwhelmed, fibroblasts and cardiomyocytes will excrete damaged mitochondria through exosomes [96]; Damaged mitochondria in neurons are selectively expelled outside the cell and then taken up and degraded by astrocytes [97]; During the differentiation process, stem cells allocate more defective mitochondria to the progenitor cells of daughter cells and more normal mitochondria to the stem cells of daughter cells [98].
There are many other pieces of evidence that do not support the mitochondrial theory of cellular aging, which are not listed one by one in this paper. In conclusion, the mitochondrial theory of aging is wrong.
• • •
Missing some Tweet in this thread? You can try to
force a refresh



