
Okay #epitwitter and #genepitwitter, let’s talk about how statistical and biological gene-environment interactions relate to each other (or not). \thread (part 1)
TL;DR 1: the distribution of a trait conditional on genotype and exposure at the population level (whether there is a statistical interaction or not) is consistent with 1,000s of possible biological models.
TL;DR 2: conversely, knowing that a gene product and an exposure or exposure byproduct physically interact at the molecular or cellular level need not say anything about what’s happening at the population level.
Hence: "The elucidation of biological interactions by means of statistical models requires the imaginative and prudent use of inductive and deductive reasoning: it cannot be done mechanically." Siemiatycki and Thomas (1981) Int J Epidemiol.
I’ll discuss definitions and intuitions and implications in a sec—but first, I want to recommend two papers that helped clarify my thinking on this. They are well worth the read. PMIDs 1999681 7327838. 


Part of the confusion around "gene environment interactions" stems from using the same words to describe different phenomenon. It's important to define what we mean by statistical and biological interactions.

Statistical interactions are easier to define, because we can write them down in maths.
Statistical interaction refers to effect measure modification of one factor by another--if the measure of the effect of exposure differs by genotype, we say there is a gene-environment interaction.

The challenge here is there are many effect measures or scales that we could use--e.g. for binary traits: odds ratios, risk differences, etc. Interaction on one scale need not imply interaction on another, and absence of interaction on one scale need not imply on others.
Consider a simple example, with a binary genotype and binary exposure. These two factors define four risk strata, and you can parameterize the four disease probabilities in many ways--here I'm showing the absolute risk and log odds scales (i.e. linear versus logit link in a GLM).

If the risk difference comparing exposed to non-exposed individuals differs by genotype (i.e. bge != 0), then we typically call that an "additive interaction" (or more verbosely: departures from additivity on the absolute risk scale).
If the odds ratio comparing exposed to non-exposed individuals differs by genotype (i.e. betage != 0), then we typically call that a "multiplicative interaction" (or more verbosely: departures from additivity on the log odds scale).
It's pretty common for folks to test for "interaction" by running a logistic regression and testing H0: betage=0. If this is non-significant, they sadly declare “no interaction” and move on.
But it’s important to realize that lack of interaction on a multiplicative scale often implies interaction on the additive scale—which can have clinical or public health implications.
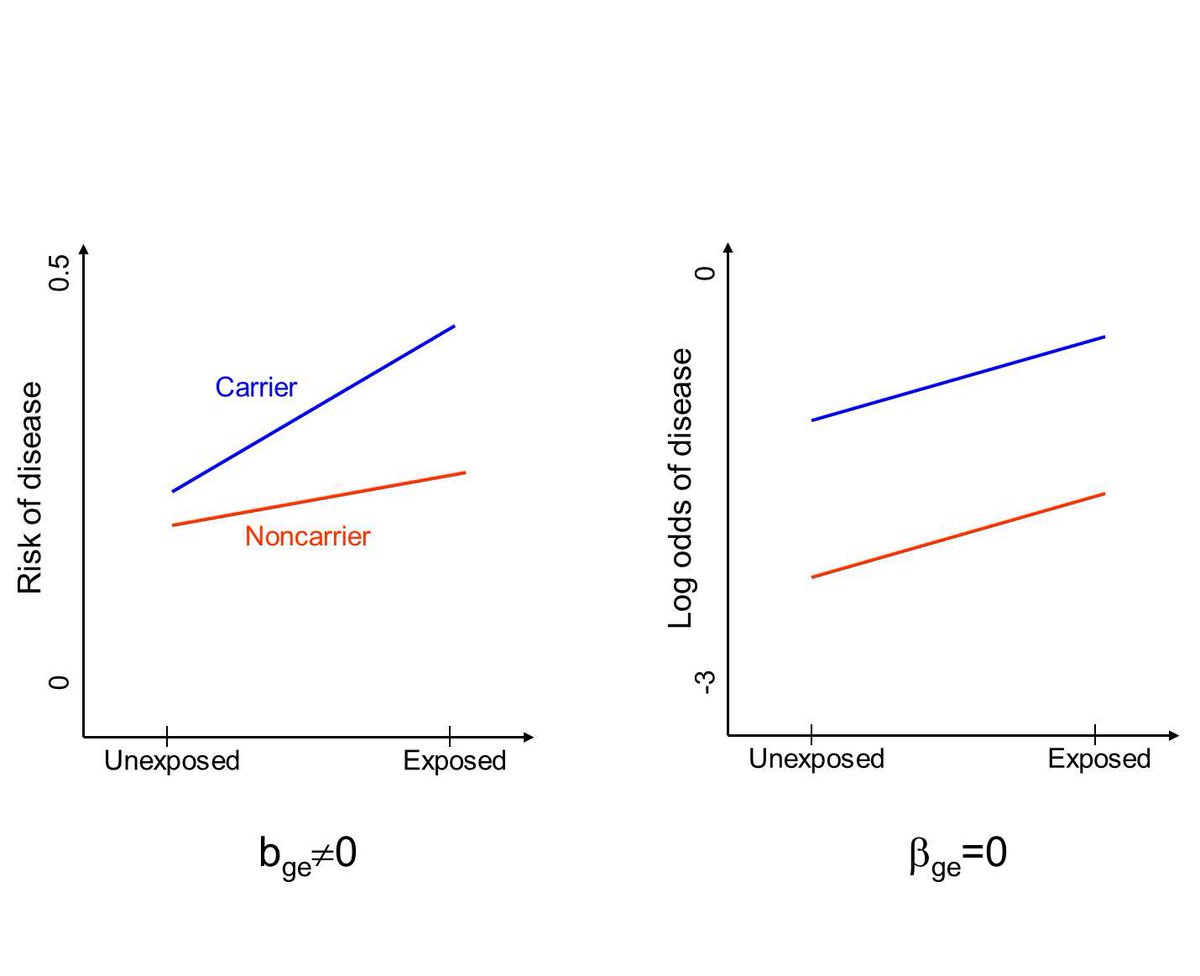
In this hypothetical case, the risk reduction from removing exposure is higher among carriers than non-carriers. If all you did was run your logistic regression to test for “interaction,” you’d miss that.

Okay, that’s statistical interaction. What about biological interaction?
This is harder to define. Most people think of this as a mechanistic interaction, where the gene product and the exposure or some byproduct physically touch.
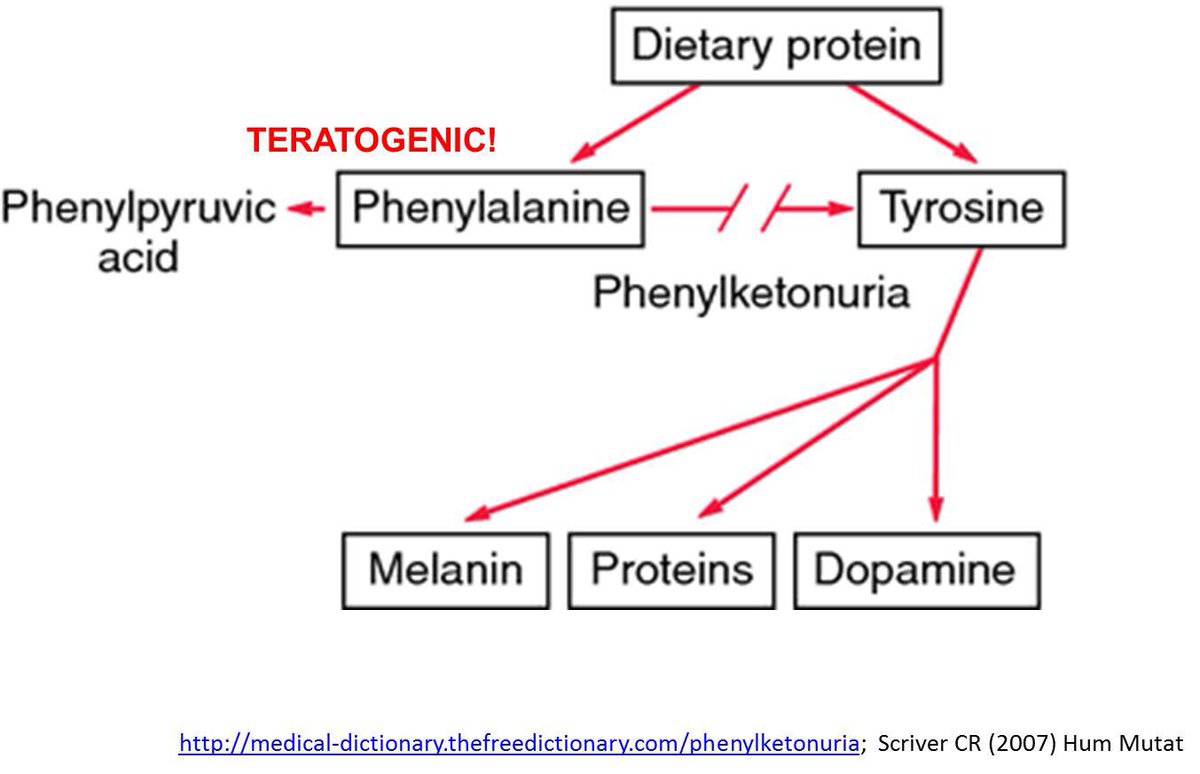
For example, folks who have a defective copy of the PAH gene cannot metabolize phenylalanine, which leads to phenylketonuria.
Another example: variants in the ADH and ALDH genes that change the efficiency of enzymes metabolizing ethanol.

(Side note: some people refer to “sufficient component cause” or “counterfactual” interactions as “biological interactions.” I do not, since most people think of mechanistic interactions when you say “biological interaction.”)
What does biological interaction have to do with statistical interaction? Hard to say, according to the Thompson and Siemiatycki & Thomas articles cited above.
The intuition behind their arguments? Even if you know something about the underlying biology--say, that the rate an exogenous exposure is detoxified differs by genotype--this need not induce a statistical interaction at the level of trait distribution in the population.
The shapes of the functions relating exogenous exposure to biologically active levels and the function relating active levels to traits are typically unknown. I may know something about the shape of g(G,X), but h(g(G,f(E)))=l(G,E) is another matter.

This does not mean biological interaction will never produce statistical interaction. You see it with phenylketonuria. You see it with alcohol, ADH/ALDH and esophageal cancer.
So if you already know how a variant changes gene function, and you know how that change will affect the exposure’s impact, you can hypothesize about what pattern you expect to see at the population level (i.e. decide what scale is relevant). To be continued...
Also, here’s the breakdown of odds of esophageal cancer, broken down by ADH/ALDH genotype and alcohol intake.

• • •
Missing some Tweet in this thread? You can try to
force a refresh






