In a new preprint, @sinabooeshaghi et al. present deep SMART-Seq, @10xGenomics and MERFISH #scRNAseq (37,925,526,323 reads, 344,256 cells) from the mouse primary motor cortex, demonstrating the benefits of cross-platform isoform-level analysis. biorxiv.org/content/10.110… 1/15 

We produce an isoform atlas and identify isoform markers for classes, subclasses and clusters of cells across all layers of the primary motor cortex. 2/15

Isoform-level results are facilitated by kallisto isoform-level quantification of the SMART-seq data. We show that such EM-based isoform quantification is essential not just for isoform but for gene-level results. #methodsmatter 3/15

Using the 10xv3 data, we also present the first cross-platform validation of SMART-seq isoform quantification (possible by analyzing transcripts with unique sequence near the 3’ end). 4/15

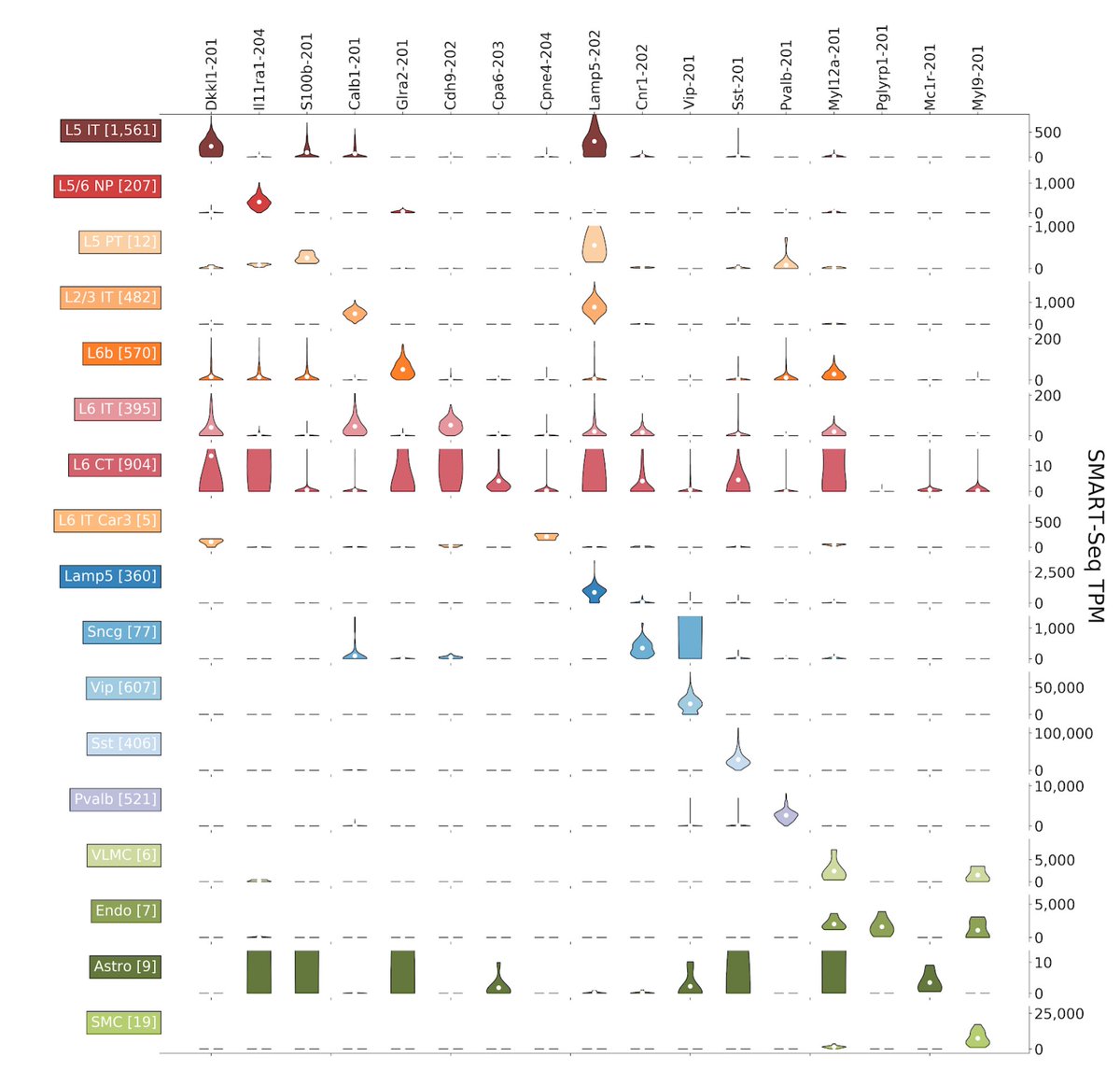
A key result is that many cell types have strong isoform markers that cannot be detected at the gene level. See the example below: 5/15

One exciting application of isoform quantification is spatial extrapolation. We show that in some cases isoform resolution can be achieved spatially even with gene probes. This is a powerful way to leverage SMART-seq for MERFISH and SEQFISH. 6/15

These results argue for a rethinking of current #scRNAseq best practices. We find that SMART-seq complements droplet-based methods and spatial RNA-seq, adding layers of important isoform resolution to cell atlases. 7/15
We recommend droplet-based high-throughput methods for cell type identification, SMART-seq for isoform resolution, and spatial RNA-seq for location information. The whole is great than the sum of the parts. 8/15

The beautiful t-SNE above is a lot cleaner than is usually the case. This is thanks to an idea of @sinabooeshaghi who made it w/ t-SNE of neighborhood component analysis (NCA) dimensionality reduction (@geoffreyhinton, Roweis et al. cs.toronto.edu/~hinton/absps/…) rather than PCA. 9/15
NCA finds a projection that maximizes a stochastic variant of the leave-one-out kNN score given cluster assignments. Intuitively, it projects so as to keep cells from the same cluster near each other, which is exactly what we want. 10/15
You might worry that NCA overfits. It doesn't. We ran a permutation test to confirm that we are seeing real structure in the data. 11/15

As an aside, we found that the t-SNE of the NCA projection preserved global structure better than t-SNE of the PCA projection (e.g., in terms of inhibitory / excitatory neuron classes). It seems that people have been confounding the performance of t-SNE (& UMAP) with PCA. 12/15
The preprint has code associated to every figure, with links directly to Jupyter notebooks hosted on @github (github.com/pachterlab/BYV…). #reproducibility #usability 13/15
All this based on amazing publicly available data from the BICCN, and this is just a preview of the whole mouse brain which is on its way. 14/15

All of this is a result of incredible work by @sinabooeshaghi who did all the analysis single-handedly. Follow him for more interesting #scRNAseq in the near future. 15/15

• • •
Missing some Tweet in this thread? You can try to
force a refresh