Hoy es el turno de #BIOINFO101 parte 2: alineamientos y filogenias, o que nos dicen las secuencias sobre el origen y la migración del coronavirus.
#hilodeciencia
#hilodeciencia
Este hilo parte de los conceptos básicos de secuencias, ADN, ARN y proteínas que vimos en la parte 1. Sino lo viste o necesitas una refrescada podés verlo acá:
Supongamos que tenemos dos secuencias cortitas de ADN: ATTG y ATTA ¿Cómo las podemos comparar?
Intuitivamente se nos puede ocurrir colocar una abajo de la otra y ver cuanto coinciden:
Intuitivamente se nos puede ocurrir colocar una abajo de la otra y ver cuanto coinciden:

Es uno de los casos más simples que vamos a encontrar porque había una sola diferencia y estaba en el final. En muchas ocasiones se producen eventos más complejos: como la introducción de una "letra" que antes no estaba (inserción) o la pérdida de una (deleción).
Si repetimos el enfoque “naive” que usamos antes tenemos 2 coincidencias entre las dos secuencias. Pero si consideramos una táctica un poco más inteligente y permitimos que las secuencias tengan huecos (o “gaps”) pasamos a tener 3 coincidencias.

Lo que hicimos en estos mini-ejemplos es lo que se conoce como *alineamiento de secuencias*. Así como alineamos una secuencia de ADN se puede hacer lo propio con ARN y proteínas.
Tengamos en cuenta que lo hicimos con una secuencia muy muy cortita. Cuando aplicamos esto sobre secuencias reales ya no podemos hacerlo “a ojo” sino que entran métodos matemáticos y computacionales en juego (o sea, ¡bioinformática!).
Veamos un ejemplo real de alineamiento, el de una proteína, la insulina. Este es un fragmento del alineamiento para el que consideré solo algunas especies de interés:

“En biología todo encuentra su sentido a la luz de la evolución” dijo alguien hace mucho tiempo y esta no es la excepción. Que todas estas especies fabriquen insulina y que todas sean tan parecidas en su secuencia no es casualidad. Es evidencia de un origen común.
Hace millones de años existió una especie, antecesora de todas las que analizamos, que ya fabricaba insulina y de que la heredamos esta característica.
Una de las claves de la evolución es la variabilidad. Las secuencias cambian de una generación a otra por motivos como la reproducción sexual (que mezcla ADN de los dos progenitores) y los errores que se producen al copiar el material genético, entre otros.
Esa variabilidad natural explica las diferencias que vemos entre las secuencias de, por ejemplo, humanos y vacas. Usualmente que una variante genética se vuelva común en una población lleva mucho tiempo, hablamos de miles o millones de años.
En comparación, los virus, y en especial los de ARN como el coronavirus, mutan MUY rápido. Y esto nos da mucha información sobre su evolución.
En el trabajo de Zhang (abril 2020) proponen que el origen del SARs-CoV-2 posiblemente está en los pangolines.
En el trabajo de Zhang (abril 2020) proponen que el origen del SARs-CoV-2 posiblemente está en los pangolines.
Zhang y colegas alinearon las secuencias de la proteína “pincho” del virus encontrado en humanos, pangolines y murciélagos.

En este caso un punto indica coincidencia y una letra indica diferencias. El alineamiento muestra una coincidencia muy alta entre el virus encontrado en humanos y en pangolines.
Es decir que para explicar el origen del virus no hace falta hacer uso de teorías conspiranoicas que hablan de que fue fabricado en un laboratorio. La evolución muestra que la hipótesis más sólida es que pasó de forma natural.
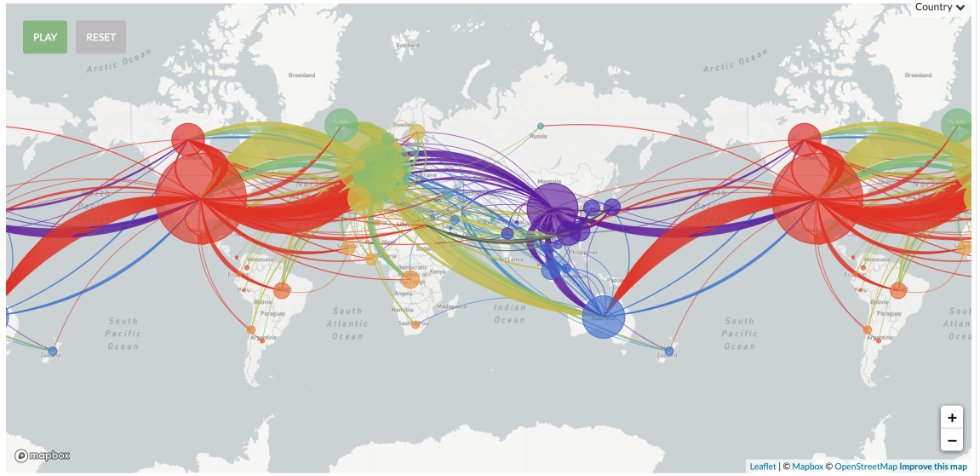
El coronavirus, a su vez, muta tan rápido que podemos no solo compararlo con otros virus sino también consigo mismo. La idea, a grandes rasgos, es que el virus una vez que pasó desde China a América (u otro lugar) fue mutando y diferenciándose del “original”.

De manera que conociendo la secuencia del virus, que es lo que en Argentina están haciendo grupos del Malbrán y otros centros, podemos hacer “vigilancia genómica”: saber de donde viene el virus que encontramos en el país.
Conocer las secuencias entonces nos permite conocer la dinámica del virus pero también podría tener consecuencias en el tratamiento. Si hay diferencias importantes entre el virus que circula en Asia y América, quizás se necesiten diferentes tratamientos.
A partir de los alineamientos de secuencias podemos armar un diagrama que las ordene, poniendo más cerca las que se parecen más entre sí. Este diagrama, que nos muestra la historia evolutiva, se llama árbol filogenético.
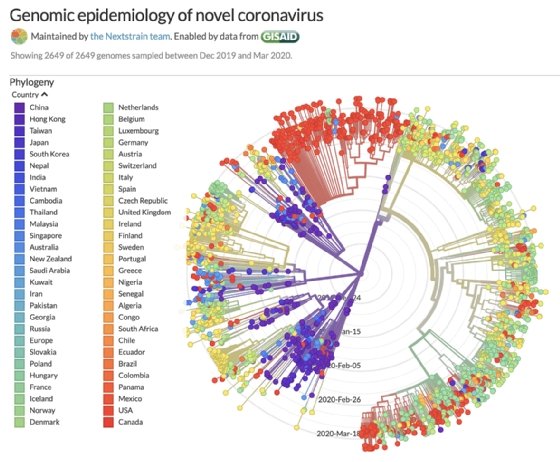
Las últimas imágenes vienen del fantástico trabajo que hacen en nextstrain.org/ncov/. En el árbol de arriba se pueden ver grupos bien marcados en las secuencias del SARs-CoV-2, correspondientes a Asia, América y Europa.
Cerrando el hilo: partiendo de los bloques que construimos en el hilo anterior avanzamos un poco más en lo que es la bioinformática hablando de alineamientos, evolución y filogenia. Y aplicamos todo esto para entender un poco más la pandemia.
Si llegaste hasta acá gracias y nos vemos la próxima en #BIOINFO101.