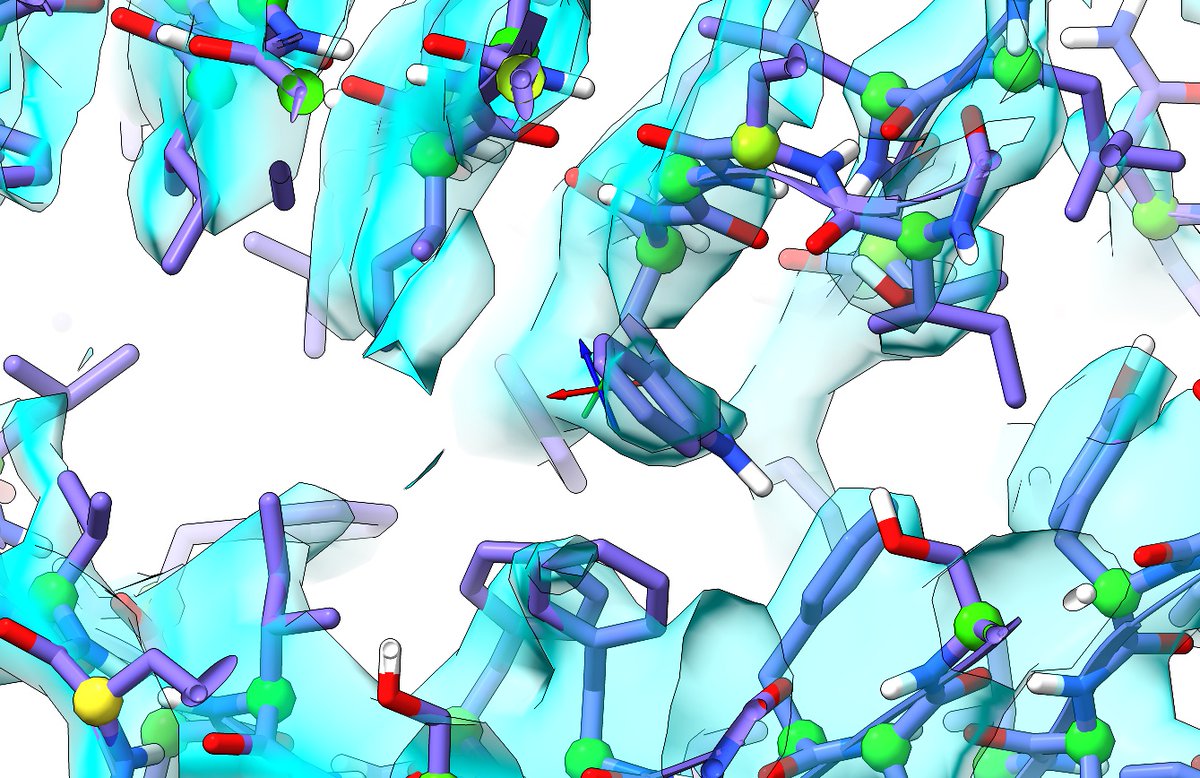
ISOLDE 1.0 is finally live! To get it, just install ChimeraX 1.0 from rbvi.ucsf.edu/chimerax/downl…, then go to Tools/More Tools..., find ISOLDE and click Install. In the thread, I'll give a quick recap of what ISOLDE is, followed by a rundown of what's new. (1/17)
So what is ISOLDE? In brief, it's an interactive environment for (re)building atomic models into medium-low-density crystallographic and cryo-EM maps using GPU-accelerated interactive molecular dynamics. That's a bit of a mouthful, so here's a video demo in the next tweet. (2/17)
This example (found this morning) shows the correction of an out-by-one error in a beta strand (residues 306-318 of 3mca chain B). Like all videos in this thread, it's an actual-speed screen capture. Key features to note: (3/17)
- Interactive MD basis (engine: @openmm_toolkit; forcefield: @ambermdprog) works to keep interactions physically reasonable;
- real-time validation of protein backbone and sidechain geometry;
- continuously updated crystallographic maps.
(4/17)
- real-time validation of protein backbone and sidechain geometry;
- continuously updated crystallographic maps.
(4/17)
The overall aim is to make it as quick and painless as possible to start from a preliminary model (from homology, auto-building or what have you), and polish it to deposition-ready standards.
(5/17)
(5/17)
An important caveat: ISOLDE is *not* designed to be an automatic package. It's built on the premise that human inspection of the result remains a key part of the model-building process, so that element should be made as painless and non-frustrating as possible. (6/17)
Anyway, on to what's new with 1.0. First: significantly improved support for when your model is not quite simulation ready (when it has residues that the MD engine doesn't recognise because they're either incomplete or not parameterised). (7/17)
This used to be, I admit, a really painful scenario. Click play, get an error after a few seconds, then try to figure out what's wrong and how to fix it. The new unparameterised residues tool finds all unmatched residues, suggests the most likely matching MD templates, (8/17)
... and fixes them to match your choice with a simple button click. If you can't find parameters for your ligand, never fear! If you have Phenix installed and available, there is now a built-in pipeline to phenix.elbow to generate the necessary AMBER params. (9/17)
Caveats: this is currently only supported for non-covalently bound ligands with no metals. On first run, this will also need to install ParmEd into your ChimeraX environment - this is automatic, but does need a compiler available (MacOS: XCode, Windows: Visual Studio) (10/17)
The new "isolde add ligand" command will add any known ligand from the Chemical Components Dictionary. A specialisation of this command, "isolde add water", adds a water molecule and automatically starts a local sim to settle it. (11/17)
Note that this is a work in progress. For large/flexible ligands it currently makes no attempt at an initial fit to the density. Even so, with judicious use of "isolde ignore" and interactive position restraints, placing these is still reasonably straightforward. (12/17)
Also somewhat experimental (but hopefully useful right now): the "isolde replace ligand" command will turn one ligand into a related one, keeping and renaming all atoms they have in common. (13/17)
One major caveat: "isolde replace ligand" is not yet chirality-aware, so using it to replace, say, a D-sugar with its L-enantiomer will not end well. If chiral centres are found in the ligand, it will print a warning to the log. (14/17)
The new ISOLDE menu also includes a handful of simple building tools that you might occasionally need: form/break disulphide, add/remove a bond, etc.. These will most commonly come up when your starting model is a PDB file without SSBOND and/or LINK records. (15/17)
Finally, in response to an overwhelming number of requests: the new "isolde step" command makes it much easier to systematically work residue-by-residue through your model. If I'm honest with myself, after adding it I can't imagine how I went so long without it! (16/17)
Anyway, please enjoy. As always, constructive feedback and suggestions are more than welcome, as are bug reports (keep 'em coming - just use ChimeraX's "report a bug" option under Help). Also, if you have tools that you think would work well with ISOLDE I'm always open. (fin)