
Our STAAR paper just appeared in @NatureGenet. STAAR performs scalable, powerful rare variant associations tests for Whole Genome Sequencing studies by using multiple in-silico functional annotations, applied to lipids in @nih_nhlbi TOPMed 30K genomes (1/)
nature.com/articles/s4158…
nature.com/articles/s4158…
Thanks go to the co-first authors (@xihaoli and Zilin Li), @zhouhufeng, @sheilamgaynor, @sunryan(on twitter), many TOPMed colleagues, including @pnatarajanmd, Gina Peloso, @cristenw, Jerry Rotter, Harvard Analysis Center colleagues of @NHGRI_GSP @bmneale and Shamil Sunyaev(2/)
WGS association analysis is challenged by a massive number of rare variants. For TOPMed Freeze 5 data of 54K genomes, 47% of variants are singletons and 16% are doubletons, <3% are common variants (with MAF>1%). >95% variants are non-coding variants (3/)
STAAR (variant-Set Test for Association using Annotation infoRmation) is a general framework for analyzing large WGS RV association studies at scale by accounting for both population structure & relatedness using linear & logistic mixed models for quantitative & binary traits(4/)
STAAR allows for two types of WGS RV analyses: Gene-centric and genetic region analyses. Gene-centric analysis groups SNVs into several coding & non-coding masks per gene: LOF, nonsynonymous, synonymous, promoter and enhancer. Region analysis uses agnostic sliding windows. (5/)
For each variant set, STAAR boosts the power of RV association tests by dynamically upweighting functional variants using multiple functional annotation scores & MAFs. Functions of a SNV are often multi-facets, & difficult to be captured by a single functional annotation score(6/
To address this, we introduce annotation Principal Components (aPCs), multi-dimensional summaries of in-silico coding & noncoding variant functional annotations. The aPCs of a SNV are the first PCs of individual in-silico functional annotations in given functional categories (7/)
Examples of aPCs include aPC-Protein, aPC-Conservation, aPC-Epigenetics, ePC-local nucleotide diversity, e.g., aPC-epigenetics is the first PC of a dozen of epigenetic scores of a SVN calculated using ENCODE & Roadmap data. Here is an aPC & individual functional score heatmap(8/) 

Annotation PCs (aPCs) are different from traditional population genetic PCs. aPCs measure individual variant’s functional annotations and are defined at the variant level, while population genetic PCs measure individual subject’s population structures at the subject level (9/)
We built FAVOR (Functional Annotation of Variants - Online Resource), an open-access variant functional annotation portal for WGS data (favor.genohub.org). FAVOR provides individual and integrated annotations that span a spectrum of variant attributes (10/)
Examples of variant functional annotations provided by FAVOR include variant categories, aPCs and other integrative scores (e.g, CADD), clinvar information, TOPMed, Gnomad & 1000G MAFs. One can do single variant, region-based & gene-based query, as well as batch submission (11/)
The current version of FAVOR includes functional annotation information for 549 millions of TOPMed BRAVO variants and indels (bravo.sph.umich.edu/freeze5/hg38/). More data will be provided in future releases.(12/)
STAAR boosts the power of the RV association tests between a variant set and a phenotype by incorporating these aPCs, other integrative functional scores, e.g., CADD, categorical functional variables, and MAFs in the STAAR test statistics using an omnibus weighting scheme (13/)
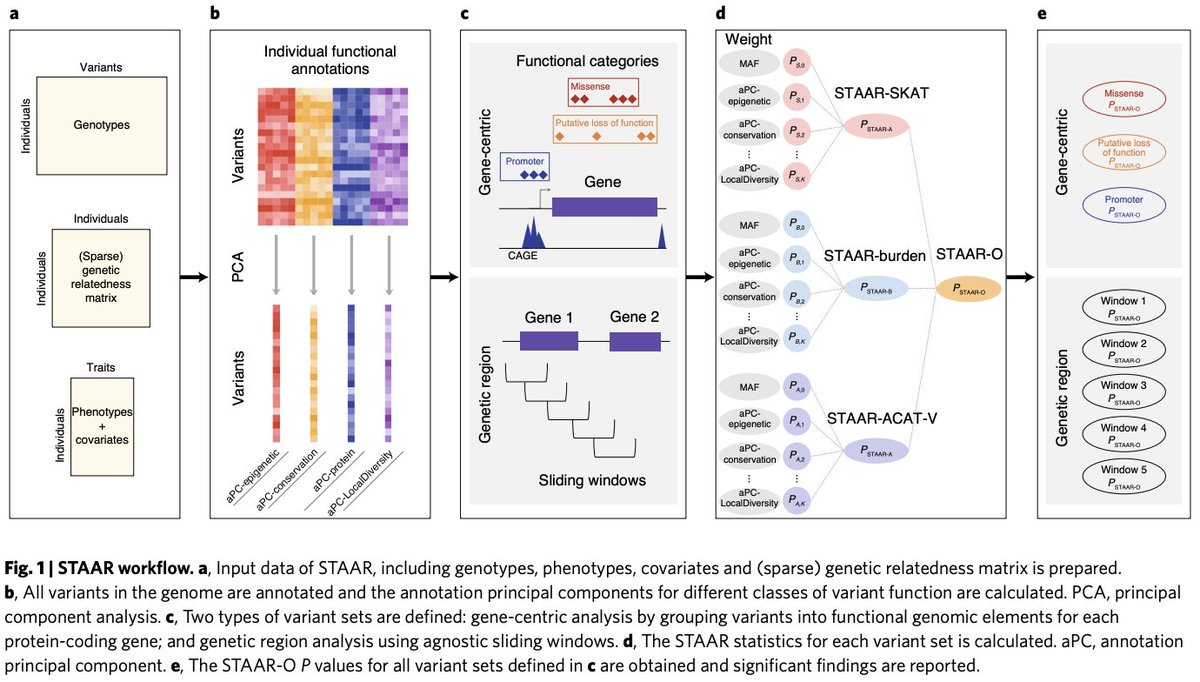
STAAR-burden, STAAR-SKAT, STAAR-ACAT-V combine the p-values of different annotation weighted burden, SKAT, ACAT-V tests using the ACAT method, respectively (14/)
STAAR-O is an omnibus test by combining p-values across different types of multiple annotation-weighted variant set tests (STAAR-burden, STAAR-SKAT, and STAAR-ACAT-V) using the ACAT method (15/)
STAAR-O is powerful when any of the incorporated variant functional annotations can pinpoint causal variants (and help boost power), and is robust to the directionality of effects and sparsity of causal variants in a variant set (16/)
In extensive simulations of various scenarios, STAAR-O achieves a significant power gain compared with conventional variant set tests weighted by MAF, such as SKAT and burden, while maintaining accurate type I error rates for both quantitative and dichotomous phenotypes (17/)
We demonstrated the use of STAAR-O in a WGS RV analysis of lipid traits using the TOPMed WGS data (12,316 discovery and 17,822 replication samples) using functional mask based gene-centric analysis and genetic region analysis using variant sets defined by 2-kb sliding windows(18/
STAAR-O outperforms existing methods, e.g, SKAT and burden tests, and identifies new & replicated conditional associations in gene-centric analysis, including with LDL-C in disruptive missense RVs of NPC1L1, and in an intergenic region near APOC1P1 in genetic region analysis(19/)


Here is a summary of RV findings of the four lipid traits (LDL, HLD, TG, TC) of the TOPMed Freeze 5 data using STAAR (20/)

Incorporating relevant tissue-specific epigenetic annotations can boost power of RV association analysis: using liver aPC as weights results in more significant RV findings for LDL (21/)

STAAR is fast & scalable for large WGS studies & biobanks by using sparse GRMs to fit the null model & scan the genome using score stats. Est computing time for UK biobank WGS analysis is 1 hr for gene-centric analysis & 25 hours for sliding window analysis if using 100 CPUs (22/

The R package STAAR can be downloaded from github.com/xihaoli/STAAR and content.sph.harvard.edu/xlin/software.… (23/)
More information about TOPMed can be found at nhlbiwgs.org. The TOPMed WGS data can be obtained from dpGap, which contains Freeze 8 data of >100K genomes



