
Given the recent advances in treating mutant RET+ cancer, I want to take this opportunity to highlight our publication in @Leukemia (2018) entitled “RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia”.
🧵 (1/13)
rdcu.be/b6yGD
🧵 (1/13)
rdcu.be/b6yGD


We performed shRNA screens using #AML cell lines and identified RET as essential kinase. We validated these findings in competition and apoptosis assays where additional AML cell lines (+ or - for RET expression) were transduced with GFP-labelled shRNAs targeting RET. (3/13)
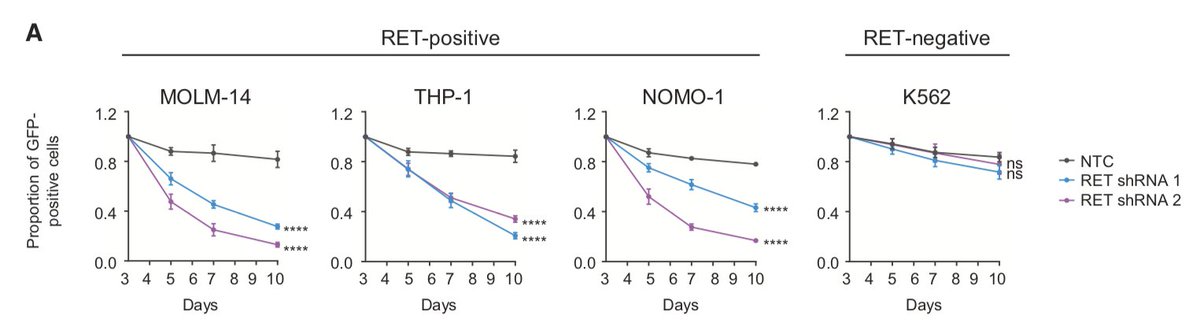
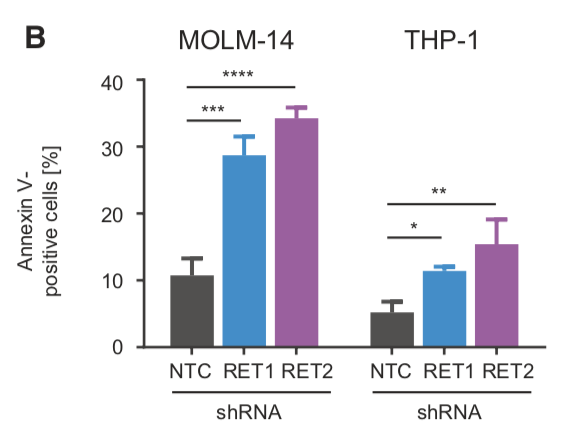
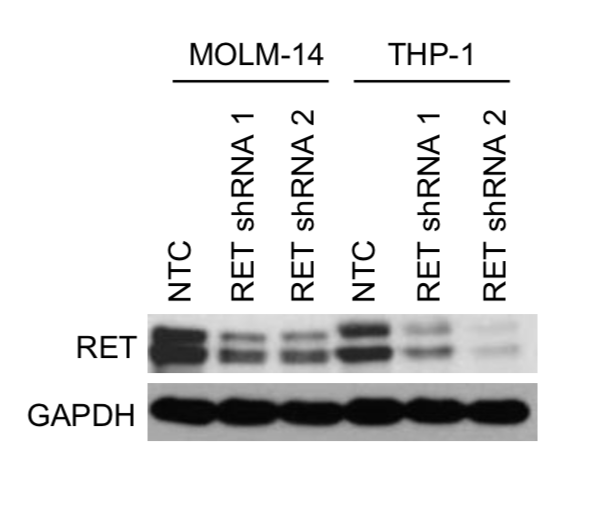
While RET is not recurrently mutated in AML, we determined that the majority (10/15) of AML cell lines and 13.5% of AML patient samples showed higher RET mRNA expression compared to normal bone marrow (BM) or CD34+ cells, respectively. (4/13)

In antibody arrays, RET was highly phosphorylated in RET+ cells, while no other RTK showed a comparable degree of phosphorylation, including FLT3, which is activated by mutation in multiple cell lines (*). RET activated the mTORC1 pathway and its effectors S6RP and p70S6K. (5/13)

mTORC1 signalling is known to inhibit autophagy, a lysosomal degradation pathway that also targets the leukaemogenic driver FLT3-ITD. In line with this, RET knockdown increased autophagic flux resulting in reduced FLT3 protein and STAT5 phosphorylation levels. (6/13)

Analysing tissue microarrays from AML patients, we found that high RET expression (immunoreactive score [IRS] 6-12) coincided with increased levels of the adaptor protein p62, which indicates suppressed autophagy, and that RET and FLT3 proteins positively correlated. (7/13)

Consistent with our observations in human cells, shRNA-mediated knockdown of murine Ret depleted cells in an in vitro competition assay and conferred a survival advantage in a mouse model of AML. (8/13)

Pharmacologic RET inhibition using vandetanib and danusertib also increased the formation of autophagic vacuoles, and cell lines dependent on mutant FLT3 displayed the highest sensitivity/lowest IC50 likely due to autophagy-mediated FLT3 degradation. (9/13)

Importantly, the RET inhibitors vandetanib and danusertib also reduced cell viability and proliferation of primary samples from FLT3-ITD+ AML patients, and co-inhibition of RET and FLT3 (crenolanib) was more effective than single treatments. (10/13)

Our results indicate that elevated expression of RET, although structurally intact, may serve as a new target for precision therapy of leukaemias that are dependent on FLT3 or, possibly, other leukaemogenic drivers whose levels are regulated by autophagy. (11/13)

This work was conducted in the labs of @claudia_scholl and @StefanFrohling at @DKFZ and @NCT_HD. Lastly, I want to thank all co-authors for their support (@YaYunCheng1125, @DanielaDiMarca1, @JanaMEllegastMD, @MilsomMick and many more who are not on Twitter). (12/13)
In case you are interested, this is the Tweet from @VivekSubbiah addressing the efficacy of selpercatinib in RET fusion+ NSCLC that prompted this thread. (13/13)

