
What started with checking some assumptions in #originsoflife and IUPAC definitions, culminated in our universal framework for autocatalysis, now in @PNASNews: pnas.org/content/early/…. The unforeseen diversity and abundance of that phenomenon convinced me to do #systemschemistry.
There are some interesting consequences that I hope to discuss in the coming few days. Today, I will outline where it all starts: definitions in chemistry and their implications. (1/n)
Like compound names and atomic weights, terminology in chemistry is critically evaluated, developed and maintained by IUPAC. Definitions in chemistry are compiled in the Gold Book: goldbook.iupac.org. (2/n)
Those definitions may not always seem “practical”: concepts like ‘catalyst’, 'chemical reaction' or 'chemical species' are defined with a degree of technical detail and nuance that requires some mastery of physical chemistry. (3/n)
Dictionaries and other sources provide an abundance of modified, simpler definitions. Many authors choose to redefine a concept like autocatalysis ad-hoc. While superficially similar, alternative definitions imply alternative properties. (4/n)
Since we want to understand ‘autocatalysis’ in chemistry, here we want concepts such as ‘catalyst’, 'catalysis', 'autocatalysis', 'reaction', 'chemical species', etc. to have properties that are consistent with their meaning and nuances in chemistry. (5/n)
Here’s a fun nuance that kept us busy: by definition, we have the flexibility to describe the same reaction using more or less reaction steps, which are themselves ‘reactions’. There is not one ‘absolute’ description of a system. (6/n)
This means that we need to be careful with how we derive conclusions about the real world from a chemical description: that conclusion should be robust to our flexibility of description. Let’s look at something we might want to look for: catalysis (7/n)
Catalysis is defined as the action of a catalyst. To qualify as a catalyst, there is a I. kinetic criterion (rate acceleration), a II. thermodynamic one (unaltered standard free energy change) and III. a stoichiometric one (reactant and product in the catalyzed reaction). (8/n) 

We may add more criteria to specify a particular flavor of catalyst. For autocatalysts, it is a stoichiometric criterion: the autocatalyst is an overall product of the catalyzed reaction (e.g. A+B->2B). (9/n)
When a reaction leads to overall production of an autocatalyst, performing it in reverse will lead to overall consumption (e.g. 2B -> A+B). The latter does not qualify as autocatalysis. Several authors refer to this case as ‘reverse autocatalysis’. (10/n)
The remaining case to fulfill the stoichometric criterion is more conventional: no net change in catalyst (e.g. A+E -> B+E). While normally redundant, it will become convenient here to distinguish this case. We will then refer to it as ‘allocatalysis’. (11/n)
The stoichiometric flavors for catalysis often combine. E.g. an allocatalyst and autocatalyst may perform A+B+E -> 2A+E. Such combinations notably come up in work on autoinduction by @DonnaBlackmond (12/n)
Such combinations are typically left implicit. For example, the autocatalytic formose reaction is routinely catalyzed by divalent metal ions and base, but leaving them out of the description is often convenient. (13/n)
This summarizes the essential properties to detect autocatalysis, and some subtleties in finding them. The paper and SI discuss some more of them. Next time: a discussion on how these properties are formalized to study autocatalysis in general (14/n)
Time for part 2, let’s take a first look at formalizing autocatalysis.
To move on, let us recall from the IUPAC definition what our formalization is concerned with:
I.kinetics, 2. thermodynamics, 3. “stoichiometry of catalytic reactions”. A little word about the last one: (15/n)
I.kinetics, 2. thermodynamics, 3. “stoichiometry of catalytic reactions”. A little word about the last one: (15/n)

By definition, a reaction is flexible. We may opt to write out an overall reaction in terms of many reaction steps. For those steps to combine into something that admits catalysis, will depend on how they are connected. (16/n)

When reactions are few in number, we may readily spot some interesting ways to combine them by listing them. When they become more numerous, it becomes practical to visualize how species and reactions are connected, and formalize those connections. (17/n)
That amounts to saying that we will study reaction networks. The way reactions and species connect shapes the structure of those networks. What the definition calls for, as we will see soon, is certain structures in a network. (18/n)
The upcoming discussion will be kept rather informal. In the paper we make it rigorous using tools, theory and insights developed for chemical networks and nonequilibrium thermodynamics. I will discuss some key references for them at the end. (19/n)
Let us write down some reactions first (left). To ensure thermodynamic consistency and check the thermodynamic criterion, each reaction is defined along with its microscopic reverse. Thereby, a choice for a ‘forward’ reaction direction is arbitrary here. (20/n)
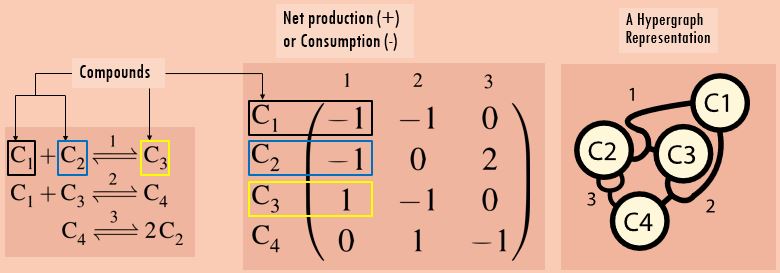
We may represent the network in a stoichiometric matrix, whose coefficients specify stoichiometry of production or consumption of participating species (middle). We may also visualize the network, e.g. by drawing a "directed" hypergraph (right). (21/n)
You may notice an issue with that matrix approach: only looking at net changes may lead to some ambiguity. We may not tell from the net change how often a species participates as a reactant and product. (22/n)
For example, A <->B, A+E <->B+E, and A + B <-> 2B would give the same net change. The capacity to distinguish between them is something we certainly need to when looking for catalysis. (23/n)
This is normally addressed by introducing more matrices to capture all coefficients. However, here the flexibility of the description works in our favor to simplify things a bit. (24/n)
The idea is that we may always pick a description where no species is simultaneously reactant and product in a single reaction step. This is readily seen by taking an ambiguous reaction and adding one intermediate to yield two successive unambiguous ones. (25/n)
We may now set a convention which we will call “nonambiguity”: we always pick a description that is not ambiguous. A stoichiometric matrix then corresponds to a unique network structure. You may surmise that catalytic structure will now imply unique matrix properties. (26/n)
The structural criterion for catalysis is not affected by how that structure is embedded in a larger network (but it is important for the kinetics!). This simplifies things: we can study the relevant structures in isolation, as subnetworks. (27/n)
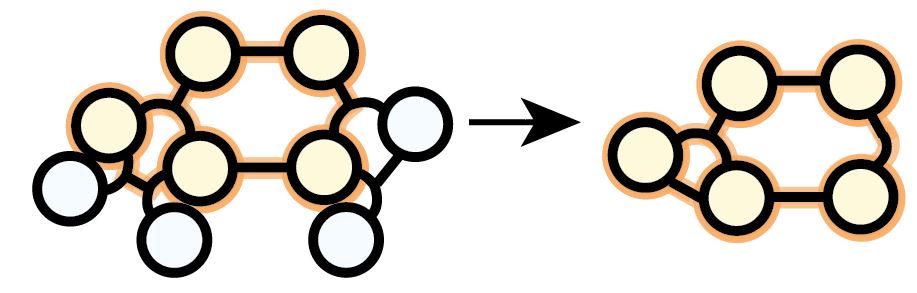
Subnetworks are obtained by removing species and reactions. Here’s what that may look like for a hypergraph representation. Sometimes these subnetworks look like regular ones. Here we may conclude something unphysical it were supposed no species were hidden. (28/n)
By the same token, a matrix becomes a submatrix. Let us do exactly that for the oversimplified Formose network shown earlier by removing C1. (29/n)
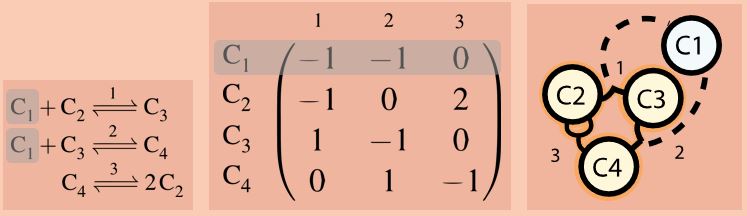
A fun way to find out more about a substructure, is to treat a submatrix as a thought experiment: reactants we remove become “fixed in concentration” and “invisible”. Allocatalysis and autocatalysis behave distinctly in this process. (30/n)
In a catalytic reaction, a reaction is performed conditional on the presence of a “substance”: the catalyst. The production of each catalyst species in the reaction must then always be conditional on other catalyst species. (31/n)

If this were not the case, we would be able to unconditionally make catalysts without their prior presence and then catalyze the reaction. The definition forbids that the reaction steps in catalysis produce such an outcome. (32/n)
Here's a concrete example of something that may ressemble catalysis in our thought experiment if we did not apply the above restriction. We retain the unimolecular reactions but get rid of half of the reactants. Like in a CSTR, species can now "appear" unconditionally. (33/n)

We refer to this conditionality on prior presence as “autonomy”. This is equivalent to the absence of empty reactions as shown before. For catalysts, it guarantees that they are all a reactant and a product at least once within a catalytic reaction. (34/n)

An interesting analogy exists with a definition for catalysis explored by @manoj_333 and the resulting siphon property for Petri nets: link.springer.com/article/10.100…. (35/n)
For allocatalysis, the catalytic reaction adds the condition that we must have an allocatalytic cycle, which performs a net reaction. Hiding reactants and products in our thought experiment make this reaction unnoticeable. (36/n)
E.g. this allocatalytic cycle would then look like an isomerization cycle. Something that distinguishes allocatalytic cycles from ‘neutral’ ones is that they only look like this in our thought experiment. (37/n)

Formally, these conditions for allocatalytic structure can be written in terms of the support and nullvectors for the stoichiometric (sub)matrix, and can be found on page 2 of the paper. Let us now mirror it with autocatalysis. (38/n)
Autonomy + stoichiometry allows autocatalytic structure to distinguish itself: autocatalysts can multiply themselves. In our thought experiment, if we only see autocatalysts, we would notice an apparent broken mass conservation. (39/n)

By definition, autocatalysts are consumed and produced, and can produce themselves overall. Application of that principle to each autocatalyst lets us infer that we can combine reactions such that each of them is produced simultaneously. (40/n)
Putting those reactions in a vector, the broken mass conservation becomes a positive vector criterion, which combines with autonomy to characterize autocatalysis. We can do a bit more, however. (41/n)
An autocatalytic structure may contain smaller substructures that would still qualify as such. Structures that admit no further smaller autocatalytic substructures are referred to as autocatalytic cores. Here we see an example: (42/n)

What can then be proven (see SI 2-3) is that minimal autocatalytic motifs have as many species as reactions, each species is a reactant in one reaction (which may be bimolecular, but only with itself), and there are no conservation laws. (43/n)
The matrix for a motif is invertible, and has several other interesting properties (see p.3). In particular, they make it possible to derive and categorize all possible core structures (SI 2-3), for which there are 5 types. (44/n)
Their general structure and explanation can be found in Fig. 2 of the text, as well as the SI. I will come back to them next time to show all the things they suddenly make possible. Let me now just give some examples of type I. (45/n)
When a description of an autocatalytic core is of Type I, the autocatalysis is using a reaction that produces the same species twice. Our simplified description of Formose does exactly that. (46/n)

But there's quite some others our there. One of my personal favorites is the autocatalytic dissolution of copper in ammonia, where we may write a characteristic type I reaction step on the copper surface: Cu + Cu(NH3)_4 2+ -> 2 Cu(NH3)_2 2+ (47/n)
Which has the advantage that it is easy to set up with kitchen chemicals. No small advantage when you need to stay home. As we'll see in a future post, there are fundamental reasons why autocatalysis becomes a lot more abundant for such multiphase systems (48/n)
In the paper (SI) we illustrate some type I cores for the reverse Krebbs cycle, in a prototypical example of autoinduction and in the GARD model. Many other ones have been realized experimentally, notably with halogens. (49/n)
To wrap up this part, some recommended reading: The 1965 Prolegomena is a delight, and concise pioneering work: link.springer.com/article/10.100… (50/n)
Feinbergs Foundations of Chemical Reaction Network Theory springer.com/gp/book/978303… is impresive by itself, but also a great place to find references to other interesting work that has remained relatively unknown (51/n)
Chemistry has pioneered many developments in nonequilibrium thermodynamics. The most recent chapter in that story, reaction networks, is promising to be its best one yet. A paper that has redefined how I see chemistry is Polettini and Esposito's: aip.scitation.org/doi/abs/10.106… (52/n)
Applying these techniques from #nonequilibriumthermodynamics to fundamental questions in #originsoflife #astrobiology #abiogenesis is a great way to extend the scientific method there. I'm excited to see it's happening more and more. (53/n)
That wraps up part 2. Next time: some interesting implications.
It’s been a while, but now it’s time for part 3. Let’s have a first look at some interesting implications at this stage of the formalization. There’s more to formalize, but network structure turns out to be most fundamental. Let’s start by making our intuitions more rigorous.
Context matters. We tend to refer to something as a ‘catalyst’ in anticipation. A fancy transition-metal carbene complex stored in a vessel will likely be used one day to actually do catalysis (e.g. olefin metathesis). While it sits there doing nothing, it does not. (54/n)
Halide ions in culinary table salt are not referred to with such anticipation. However, examples where halide ions perform catalysis are numerous (e.g. racemization catalysis). (55/n)
The gold book definition is based on contextual information about the system. That context is part of the reaction network structure. (56/n)
Calling the contents of a vessel ‘catalyst’ based on its label is a ‘loose’, alternative usage of the term. It tells us something about cultural context. That’s perfectly reasonable: many situations in life are conveniently addressed without being specific or consistent. (57/n)
In this situation being specific is convenient: we’re deriving conclusions implied by a specific set of properties that chemists around the globe have agreed on to describe a phenomenon common to all branches of chemistry. (58/n)
Let’s pause for two tweets to appreciate what standardization has done for chemistry. Below is a list (see: iupac.org/who-we-are/our…) of what had to be addressed when that process started >100 years ago. The final one, an "inverse replication crisis" is an interesting surprise.

Standardization facilitates the sharing of insights. Here’s a recent table of 1-electron radical reduction potentials in water: degruyter.com/view/journals/…. Chemists with access to the techniques and expertise to measure these are far outnumbered by the chemists that benefit from it.
We may have preferences for labels, but if you show me an exciting result a different label will not alter my excitement. For alternative definitions of catalysis, that simply means that I’ll have to do some extra work to figure out which properties you were looking at. 59/m
Alternative properties may capture particular types of catalysts, or something very different. It could even capture something that transcends catalysis, a generalization. Some variations haven’t acquired Gold-Book status, but we can already talk about their properties! 60/m
I find short quotes to be good mnemonics. Here’s what an Irish Poet suggested to transcend catalysis: “To define is to limit.” Our discussion on catalysis is restricted to those properties that define it, which simultaneously need to apply. 61/m

Heterogeneous catalysis would be more restrictive than catalysis: there’s an additional criterion. To be less restrictive, we can simply do the opposite. We transcend catalysis when we talk about a subset of the properties that define catalysis. 62/m
A motivation: here’s a teaser video illustrating exactly that (). The restoration of a Tungsten filament in a halogen lamp due to the halogen cycle. A fun daily-life example how dissipative coupling changes chemistry. 63/m
In an incandescent lamp, the hot filament slowly sublimes, and deposits on the bulb. The wikipedia page has some nice illustrations (en.wikipedia.org/wiki/Incandesc…). The one I also show here uses a carbon filament. 64/m

That filament became a lot more durable and efficient when other materials started to be used for it, like Tungsten. The history of my hometown, Eindhoven, is intimitaly tied to the production of such bulbs by Philips. 65/m
If we want to get much light out of the filament, we want to make it hot. Tungsten may boil at 6203 K, but that doesn't keep it intact. Similar to vapor pressure being sufficient to eventually evaporate a puddle of water without heating it to 373 K. 66/m
Overly simplified (here's more detail: tinyurl.com/y2k7zp97, let me know if you know a good book!), the halogen lamp can restore that. In the linked example that happens by forming WOX2, which preferentially dissociates near the hot filament to restore O and 2 X. 67/m
If we imagine the process in the motivating video to be that (tinyurl.com/y3tdt8u8), we see an analogy: O and X atoms facilitate a process of depositing tungsten from bulb to filament, but are themselves not consumed, similar to allocatalysis. 68/m
Here's why it's not good old allocatalysis: the thermodynamic criterion. The hot filament gives us net transport of tungsten to the bulb. The halogen cycle goes the other way around. If we need them to be thermodynamically equivalent, they must go in the same direction. 69/m
In fact, thermo gives much stronger restrictions. But suffice to say, it's clear that the thermodynamic equivalence property (marked in red) has to go to capture this phenomenon: 70/m

So how is the facilitated transport different? That's what the temperature gradient does for us. We're not simply displacing Tungsten, we're displacing heat. That by itself would suffice to make the thermodynamic balance intrinsically different. 71/m
And that's the crux: we're now dealing with a phenomenon where we intrinsically have an issue when comparing alternatives in the way 'uncatalyzed' and 'catalyzed' reactions can be compared. The process itself now has bearing on that thermodynamic balance. 72/m
And that takes us to a more general point: if we just drop the thermo criterion and give that concept a name 'Y', 'Y' now describes, by definition, everything we call catalysis, but in addition now also captures cases that transcend it. 73/m
Now that we've gone through that let's take a quick look at some examples we already know that do exactly that. 74/m
• • •
Missing some Tweet in this thread? You can try to
force a refresh


