In new work, we show a human coronavirus evolves to escape neutralization by antibody immunity (biorxiv.org/content/10.110…). Specifically, we studied the historical evolution of the common-cold CoV-229E to learn how #SARSCoV2 might evolve & if we might need to update vaccines. (1/n)
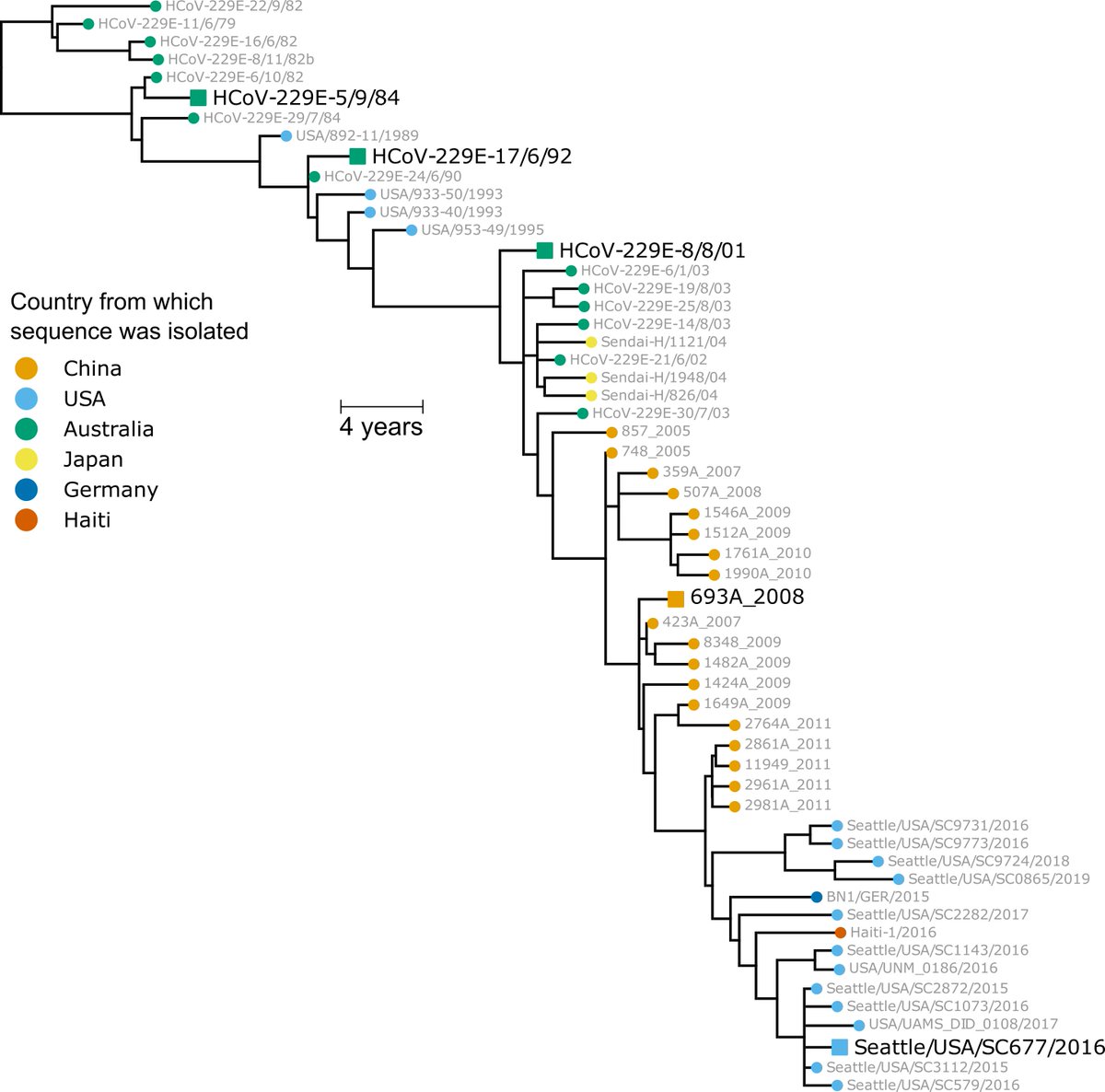
We first built a phylogenetic tree of CoV-229E evolution from 1984 to the present, and experimentally reconstructed the spike from viruses at 8 year intervals (1984, 1992, etc; see large black strain names in tree below). (2/n) 
Next we tested how well human sera collected shortly after 1984 neutralized each viral spike. Below is serum from 26 yr old collected in 1985: it neutralizes 1984 virus well, but 10-fold less activity against 1992 virus & no activity against viruses after 2008. (3/n)
Sometimes, the loss of neutralization of "future" evolved CoV-229E virus is even more dramatic. Below is serum collected in 1990 from a 28 yr old that neutralized 1984 virus very well, but has no activity against any viruses more recent than that! (4/n)
These results show that the coronavirus is evolving antigenically, so immunity elicited against older CoV-229E is eroded by mutations in spike. For instance, this serum collected in 1995 neutralizes viral spikes from before then, but has reduced activity against new spikes. (5/n)
In contrast, "modern" serum collected from adults in 2020 tends to neutralize all historical viruses (see below for example), suggesting antibody immunity itself is durable: the problem is viral evolution that escapes antibodies to older viruses. (6/n)
We did additional experiments suggesting much of the antigenic evolution is in the spike's RBD, which is the most evolutionarily variable part of the CoV-229E spike, especially in receptor binding loops (see below & paper for more details). (7/n)
Why is spike antigenically evolving so fast given that coronaviruses have lower mutation rates than other RNA viruses such as flu? Well, mutation rate is only one part of evolution, which also depends on how selection acts on effects of mutations. (8/n)
Unfortunately, CoV spike RBD is mutationally tolerant (see @tylernstarr's deep mutational scanning: sciencedirect.com/science/articl…) & selection can strongly favor mutations in seasonal CoV spikes (see Kistler & @trvrb: biorxiv.org/content/10.110…). This enables antigenic evolution. (9/n)
What does this mean for #SARSCoV2 immunity? First, need to emphasize that our work was done in CoV-229E, which is a *different* human CoV. Nonetheless, there is lots of evidence antigenic mutations occur in #SARSCoV2 too (citations in Tweet 13 below). (10/n)
But people should not be alarmed. Human immunity is polyclonal, so even in worst case it would take years to get enough viral mutations to fully escape. Furthermore, even residual immunity to antigenically evolved viruses could reduce disease severity (this is unknown). (11/n)
Furthermore, leading vaccines to #SARSCoV2 using cutting-edge approaches (eg, mRNA) that should make it easy to update spike sequence if there is evolution. So for this reason, we need to carefully monitor virus for antigenic evolution. (12/n)
This is why many labs (eg, ours, @vsv512 @PaulBieniasz @theodora_nyc) have been mapping which #SARSCoV2 mutations reduce antibody and serum neutralization (eg, elifesciences.org/articles/61312, biorxiv.org/content/10.110…, biorxiv.org/content/10.110…, sciencedirect.com/science/articl…). (13/n)
This is important! If we identify possible antigenic mutations ahead of time, then if #SARSCoV2 evolves to escape immunity like CoV-229E, we can see it happening--and if needed vaccines could be periodically updated as is already done for influenza. (14/n)
Another hopeful thing: we found some people had immunity that was resistant to viral evolution. For instance, serum of 35 yr old below neutralized CoV-229E from 2 decades later. If we learn what makes some immunity evolution-resistant, maybe we can better elicit it. (15/n)
This work was led by @eguia_rachel and @khdcrawford in our group.
Great collabs, including @GreningerLab. Although >2e5 #SARSCoV2 seqs, few CoV-229E seqs in last decade almost all from @GreningerLab @UWVirology. By sequencing "less popular" viruses, they enabled our study (16/n)
Great collabs, including @GreningerLab. Although >2e5 #SARSCoV2 seqs, few CoV-229E seqs in last decade almost all from @GreningerLab @UWVirology. By sequencing "less popular" viruses, they enabled our study (16/n)
I forgot to include image of serum from 35 yr old that had immunity that is more resistant to viral evolution. Here it is, see how this sera collected in 1986 neutralizes viruses from two decades later. Ideally, a vaccine would elicit sera like this!
• • •
Missing some Tweet in this thread? You can try to
force a refresh