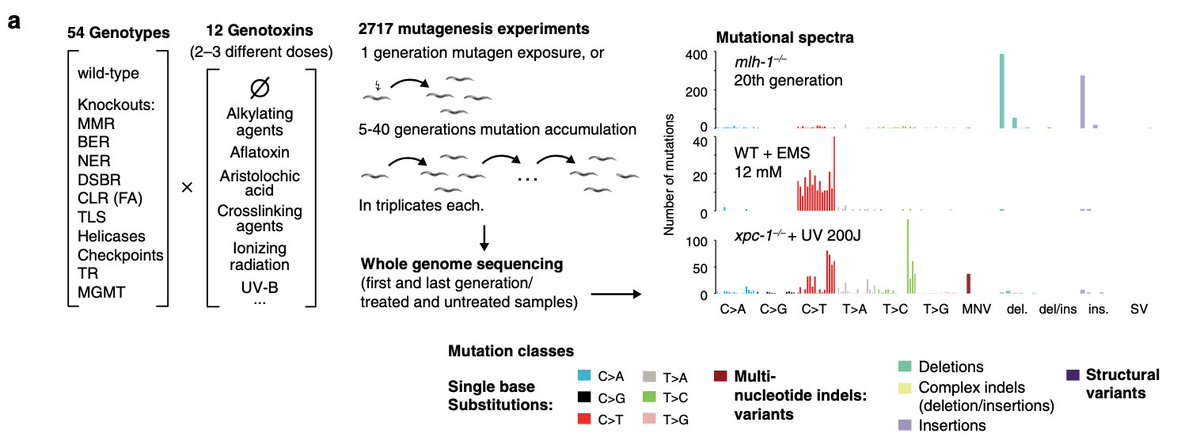
Did the new SARS-CoV-2 B.1.1.7 lineage spread during the English national lockdown? Rising numbers and estimated higher R value suggest so. Together with our colleagues from COG-UK we took a closer look. >> virological.org/t/lineage-spec… 

Fitting lineage-agnostic daily PCR test and viral genome data from COG-UK to 382 local authorities we find evidence that B.1.1.7 has spread in a staggering 200/246 of affected LTLAs during the November lockdown (R>1) while at the same time other lineages contracted (R<1). >>

The evidence is therefore overwhelming that B.1.1.7 was repeatedly capable to proliferate under lockdown measures sufficient to suppress other SARS-CoV-2 lineages. B.1.1.7 spread was not an isolated event of general failure of viral containment (both R>1). >>
Control of B.1.1.7 will therefore require stricter measures than applied during the November lockdown as noted and modelled by others. >> cmmid.github.io/topics/covid19…
As it emerges that B.1.1.7 has already spread to many other countries, everything possible should be done to contain it in its roots. Failure to stop this variant now will mean long and strict lockdowns one or two months later otherwise. >> theguardian.com/world/2020/dec…
Many thanks to @harald_voeh, @jcbarret, @imartincorena @CovidGenomicsUK and many other colleagues from @sangerinstitute and @emblebi for their hard work over the last weeks and months.
To address a few questions that came up. Some people wondered whether a lockdown would select for the new strain since it emerged during this period. In the weeks after lockdown was lifted, the spread of the new lineage was even faster than during lockdown. >>
Or put differently: if there had been no lockdown the relative proportions would about the same (this is defined by the fold change in R values), but there would be many more cases of both lineages. >>
Other people wondered about whether B.1.1.7 case numbers increased only in certain areas, possibly because of behavioural differences. Our analysis shows that B.1.1.7 case numbers increased approximately 3-5 fold during lockdown in nearly every LTLA. >>
In the South East this increase had a noticeable effect on case total because of the high B.1.1.7. prevalence (50% in some areas) at the beginning of lockdown. In other (most) areas with only a few % of B.1.1.7 this growth was masked by the decline of other lineages of ~50%.
• • •
Missing some Tweet in this thread? You can try to
force a refresh