
Hervorragend systematischen Daten aus BaWü zeigen: Mutanten-Anteil bereits bei 12% - und das im Schnitt über die letzten 7 Meldetage.
Die 6% aus dem @rki_de-Bericht sollte man schnell als veraltet abheften, lieber hierauf schauen.
Transparente Berechnung: docs.google.com/spreadsheets/d…
Die 6% aus dem @rki_de-Bericht sollte man schnell als veraltet abheften, lieber hierauf schauen.
Transparente Berechnung: docs.google.com/spreadsheets/d…
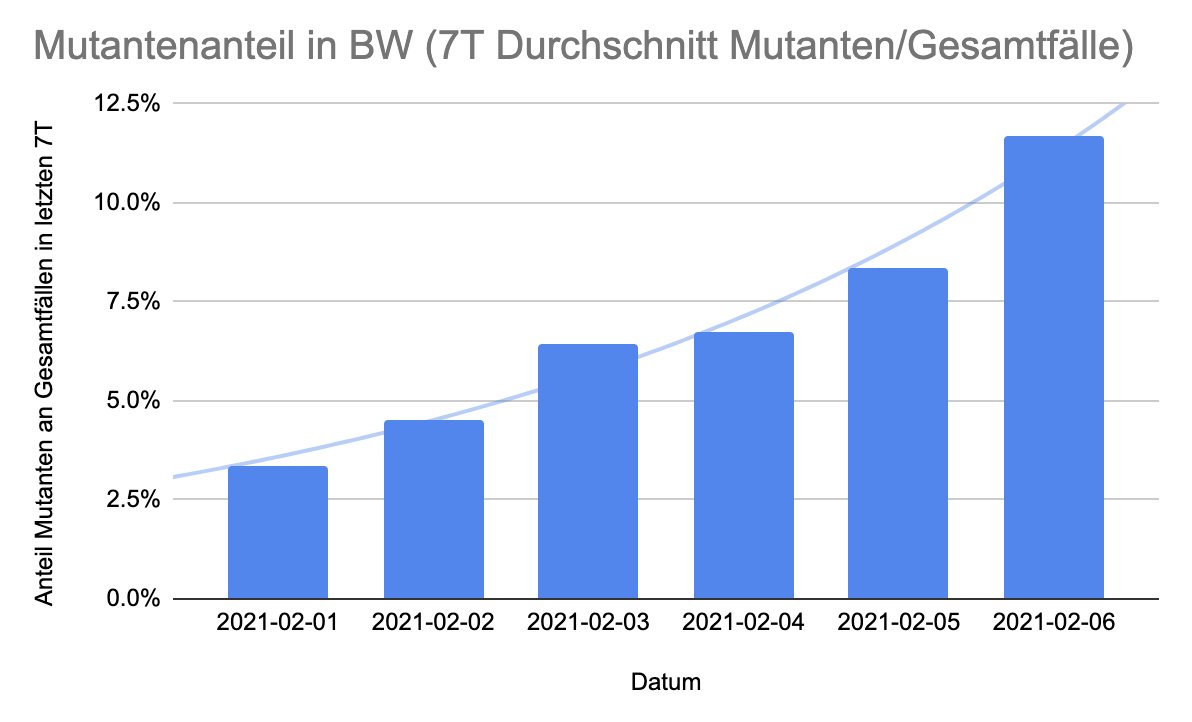
Auch die Entwicklung der Zahl der Mutantenfälle wächst exponentiell (also nicht nur der Anteil).
Trotz Rückgang der Fallzahlen steigen die Zahlen der Mutanten - wie man das bei einem R von ca. 0,9 des Wildtyps auch erwarten würde. R der Mutanten dann bei ca. 1,3. 2/
Trotz Rückgang der Fallzahlen steigen die Zahlen der Mutanten - wie man das bei einem R von ca. 0,9 des Wildtyps auch erwarten würde. R der Mutanten dann bei ca. 1,3. 2/

Warum sind die BaWü-Zahlen so wertvoll? Weil dort alle Fälle auf Mutationen untersucht werden. Sehr weise von @RegierungBW um einen besseren Überblick über die Lage zu bekommen - und nicht von veralteten, punktuellen @rki_de-Zahlen abhängig zu sein.
swr.de/swraktuell/bad…
swr.de/swraktuell/bad…
Warum steigende Zahl der Mutanten-Fälle ein großes Problem ist, zeigt dieser Artikel aus der @SZ
Nur, dass der Anteil der Mutante in Realität schon höher ist als auf dem Bild (also mehr Rot).
Wichtig: Annahme der Grafik ist, dass KEINE LOCKERUNGEN kommen.
Nur, dass der Anteil der Mutante in Realität schon höher ist als auf dem Bild (also mehr Rot).
Wichtig: Annahme der Grafik ist, dass KEINE LOCKERUNGEN kommen.
https://twitter.com/c_endt/status/1357750987029643268?s=20
Hier die gleiche Grafik mit den neuesten Daten, wurden gerade eben von @MSI_BW veröffentlicht.
Jeden Tag werden die Zahlen belastbarer, mit geringeren statistischen und systematischen Fehlern.
stm.baden-wuerttemberg.de/fileadmin/reda…
Jeden Tag werden die Zahlen belastbarer, mit geringeren statistischen und systematischen Fehlern.
stm.baden-wuerttemberg.de/fileadmin/reda…
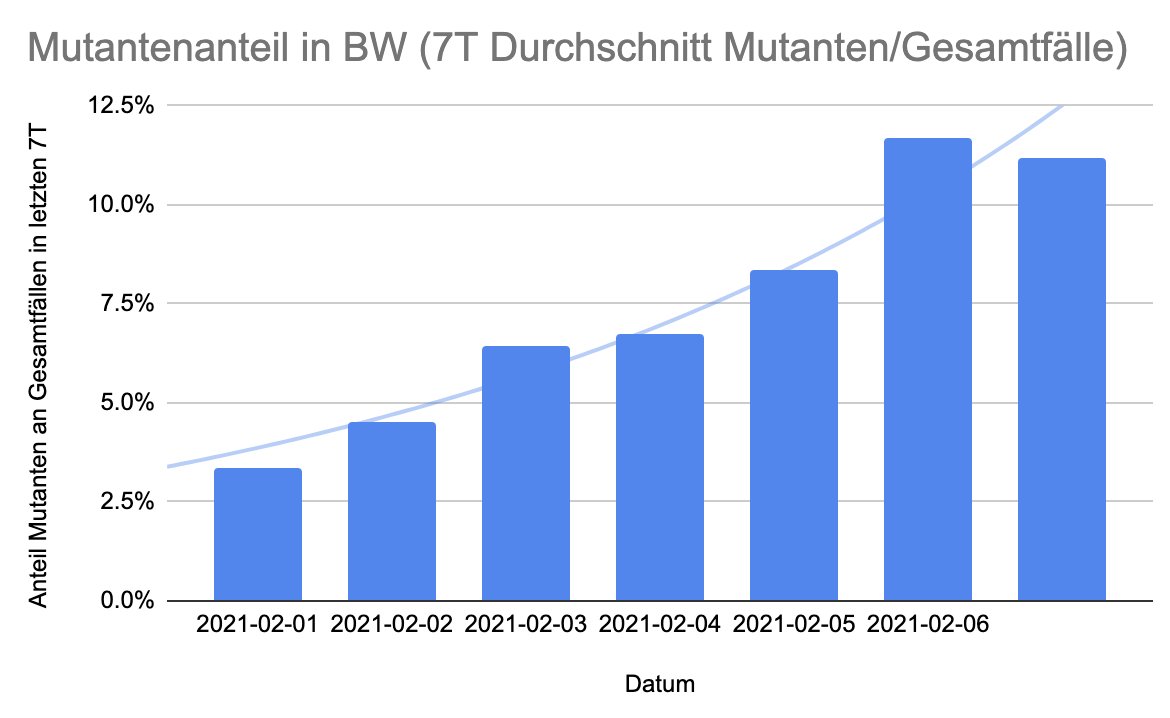
Es gibt neue Daten, Wachstum flacht ab, vermutlich wegen Nachholeffekt, es wurden ursprünglich mehr Proben getestet, als an einem Tag reinkommen sollte, deshalb Überhöhung.
https://twitter.com/CorneliusRoemer/status/1358879968403591169?s=20
• • •
Missing some Tweet in this thread? You can try to
force a refresh







