
⚙️Methods⚙️ paper📜 on RADseq
This one is for you if you think your RADseq data is 🗑️, & you feel 😭/😥 about it!
besjournals.onlinelibrary.wiley.com/doi/10.1111/20…
But before, shoutout to @MaurstadMarius who shares first-authorship w/ me, after teaching himself Unix as an undergraduate🧐- impressive!
This one is for you if you think your RADseq data is 🗑️, & you feel 😭/😥 about it!
besjournals.onlinelibrary.wiley.com/doi/10.1111/20…
But before, shoutout to @MaurstadMarius who shares first-authorship w/ me, after teaching himself Unix as an undergraduate🧐- impressive!
This paper begins with this dataset on marine ghost-worms ( peerj.com/articles/10896/ ).
We had >90% missing data across the dataset, results that were impossible to make sense from (PCA/trees), and issues just getting a reasonably high number of high-quality variants...
We had >90% missing data across the dataset, results that were impossible to make sense from (PCA/trees), and issues just getting a reasonably high number of high-quality variants...
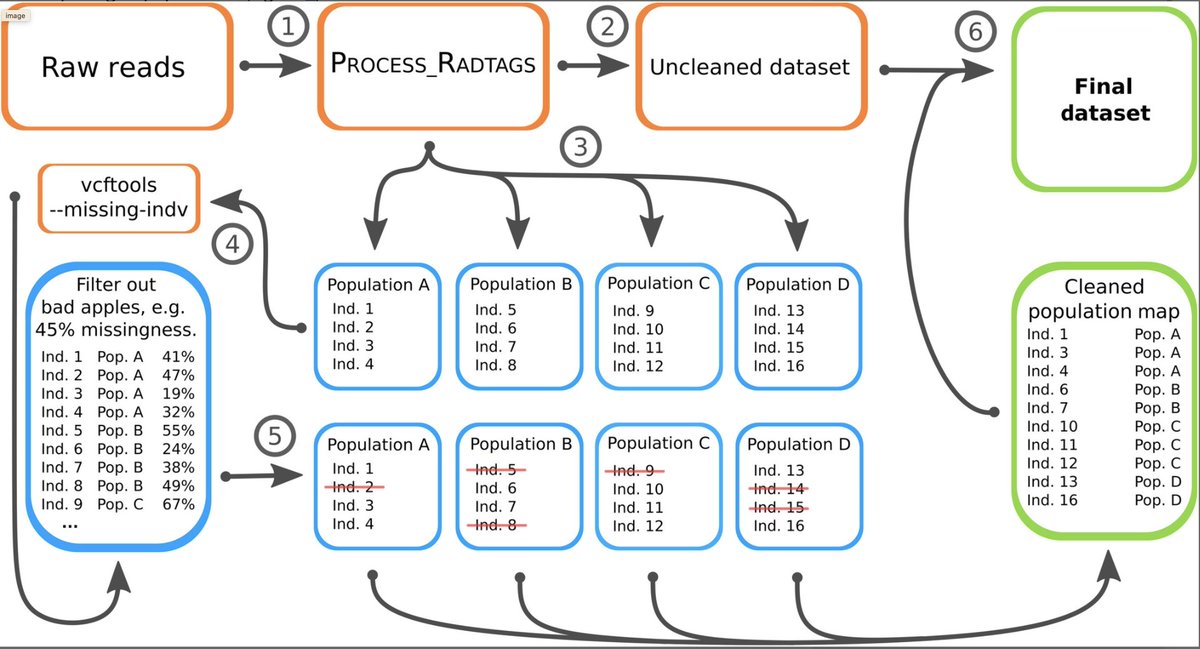
In a conversation w/ @ncrochette, he suggested I should just run stacks on a population-by-population level and explore what was happening. I noticed:
1. ≠ populations had very ≠ nr. of variants;
2. some individuals at the population level had tons of missing data (bad apples)
1. ≠ populations had very ≠ nr. of variants;
2. some individuals at the population level had tons of missing data (bad apples)

Why would specimens have tons of missing data at the population level?
One expects individuals on a population to share the majority of enzyme cut-sites. Maybe something to do with library-preparation/sequencing/DNA quality? Since ...
One expects individuals on a population to share the majority of enzyme cut-sites. Maybe something to do with library-preparation/sequencing/DNA quality? Since ...
... when dealing with various species, divergence is expected to correlate with the loss restriction sites. An increase of divergence translates to more "allele dropout" (i.e. missing data); but this is not expected at the population-level.
What we noticed is that by removing these individuals with tons of missing data, and then reassembling the dataset without them ... we obtained a much-improved dataset!
After testing this in 3 other datasets, we generally confirmed these results.
After testing this in 3 other datasets, we generally confirmed these results.

In the paper, we explore the impact of
1. removing random samples;
2. removing "bad apples";
3. removing "bad apples" in different parts of the stacks' pipeline;
&
4. explore data with PCA;
5. explore "properties" of the SNPs kept and removed (before & after removing samples)
1. removing random samples;
2. removing "bad apples";
3. removing "bad apples" in different parts of the stacks' pipeline;
&
4. explore data with PCA;
5. explore "properties" of the SNPs kept and removed (before & after removing samples)

... We conclude that this process will benefit non-model datasets by
1. increasing number of loci in the data; &
2. decreasing missing data.
One particular advantage is that we do not find evidence of removing a particular "class of loci". This is:
1. increasing number of loci in the data; &
2. decreasing missing data.
One particular advantage is that we do not find evidence of removing a particular "class of loci". This is:
A. If you are very strict in your filters, you may keep only very conserved loci;
B. If you are very liberal, you may have may pass down artefacts on your final dataset.
Both A and B have been shown in RADseq papers, which we cite and discuss.
B. If you are very liberal, you may have may pass down artefacts on your final dataset.
Both A and B have been shown in RADseq papers, which we cite and discuss.
We therefore think we came up with a simple method to "clean-up" datasets that may suffer mostly from library-building, sequencing and DNA-quality issues - which would explain "allele dropout" at the population/species level.
Big shoutout to @ncrochette @jcatchen @arcolon14 @RayamajhiN for comments and their god-tier expertise and very constructive comments on RADseq; @fez_nhm for actually suggesting we should turn this into a paper, and helping analyzing the data.
& @MaurstadMarius for being motivated with bioinformatics and doing tons of stacks' run;
That's all folks :)
That's all folks :)
• • •
Missing some Tweet in this thread? You can try to
force a refresh



