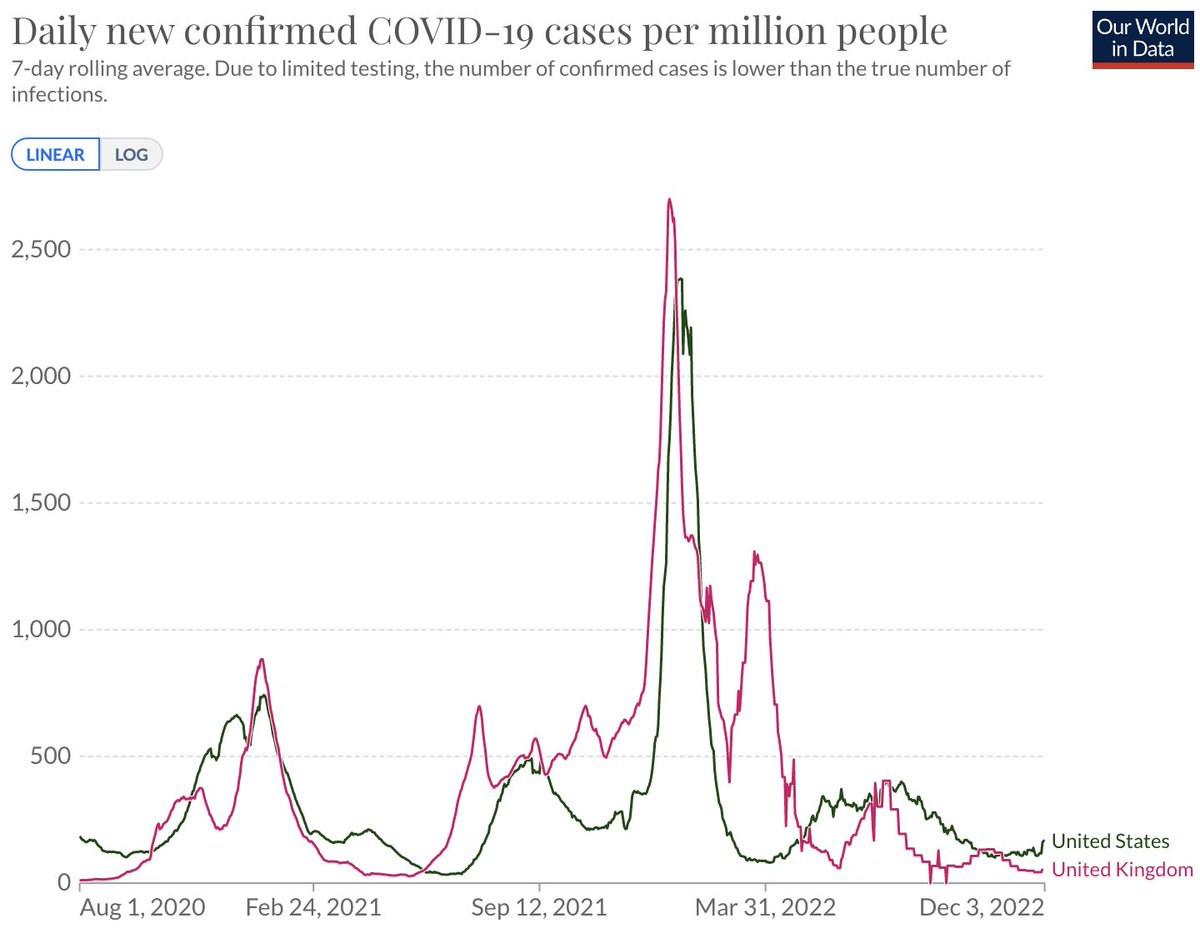
New (not yet peer-reviewed) work by Katie Kistler and @huddlej in the lab assessing adaptive evolution in SARS-CoV-2 across the viral genome. 1/12
biorxiv.org/content/10.110…
biorxiv.org/content/10.110…
We measure adaptive evolution by correlating mutations in different regions of the genome with growth of clade frequency. For this, we use a viral phylogeny of ~10k genomes sampled equitably through space and time across the pandemic (nextstrain.org/groups/blab/nc…). 2/12 

If mutations to a region result in fitter viruses, clades bearing these mutations should expand more rapidly. We find that the S1 domain of spike accumulates protein-coding (nonsynonymous) changes rapidly and that clades with more S1 mutations tend to grow in frequency. 3/12
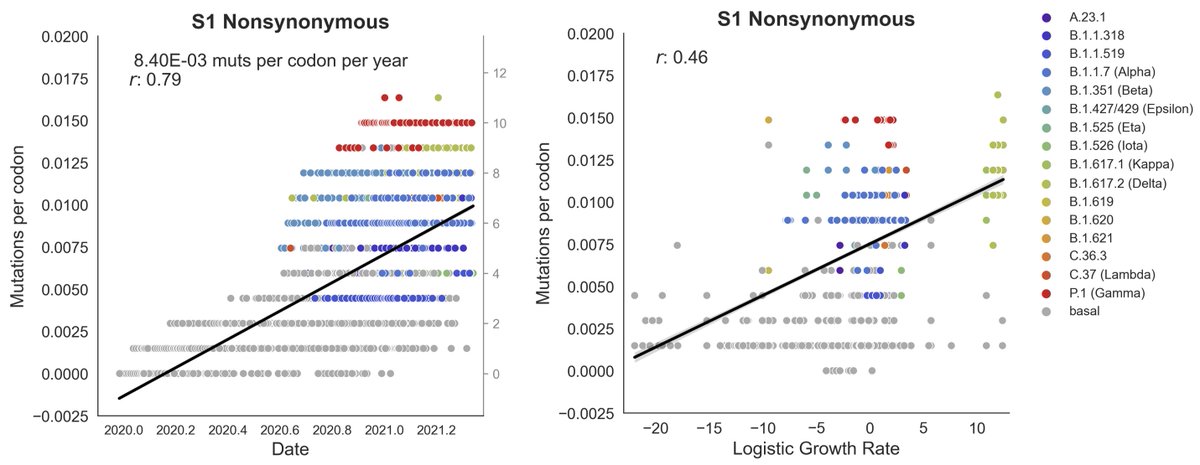
As a control we looked at non-protein coding (synonymous) mutations in S1 and nonsynonymous mutations in the RdRp polymerase. In both cases we fail to observe a significant correlation with clade growth. 4/12
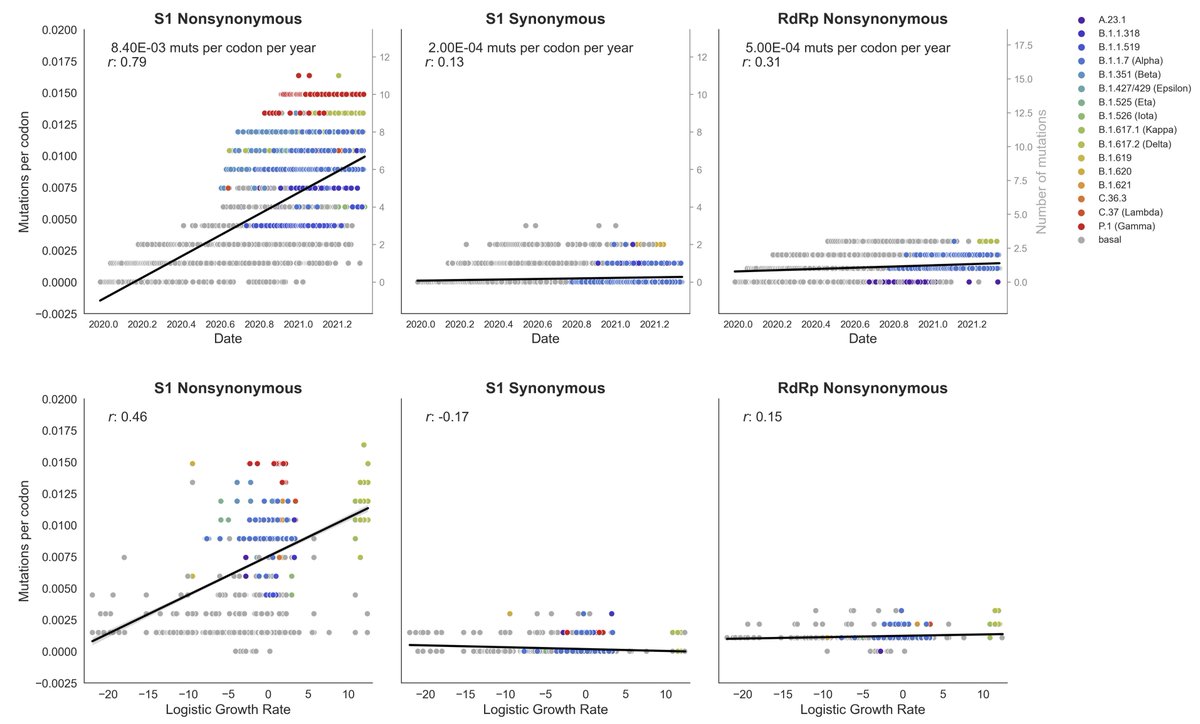
Across the genome, we see the highest correlation in the S1 domain of spike, but find weaker (though statistically significant) evidence of adaptive evolution in Nsp6 and ORF7a. 5/12

Focusing on S1, we calculate a common metric called dN/dS that compares nonsynonymous mutations to synonymous mutations. We find that dN/dS in S1 increases through time during the pandemic with the most recent timepoint showing dN/dS of ~2.1. 6/12

This is a fast pace of adaptive evolution and it's rare to observe such a strong signal. HA1 in influenza H3N2 as the canonical example of an adaptively evolving viral protein shows dN/dS of ~0.4, or about 5 times lower than what's currently being observed in SARS-CoV-2. 7/12
Further observations show that nonsynonymous mutations in S1 cluster along the phylogeny quantifying the anecdotal observation that variant viruses often have multiple mutations occurring all together. 8/12

We also observe convergent evolution in individual mutations and identify a subset that occur repeatedly in parallel and when occurring are associated with clade growth. This highlights spike mutations 95I, 452R and 484K as well as a 3 amino acid deletion in Nsp6. 9/12

Most of this rapid pace of evolution is likely due to adaptation to a new host, but in general, this suggests to me that the S1 domain of spike in SARS-CoV-2 is a readily evolvable domain. 10/12
Circulating mutations like 484K partially escape from antibody responses and although I'd anticipate the pace of evolution to slow as the virus becomes endemic in the human population, I would also expect relatively rapid antigenic drift, just given this data. 11/12
We'll of course have to wait to see what unfolds, but I would, at this point, suspect an influenza H3N2-like process of antigenic drift and necessarily frequent vaccine updates in the upcoming years. 12/12
• • •
Missing some Tweet in this thread? You can try to
force a refresh