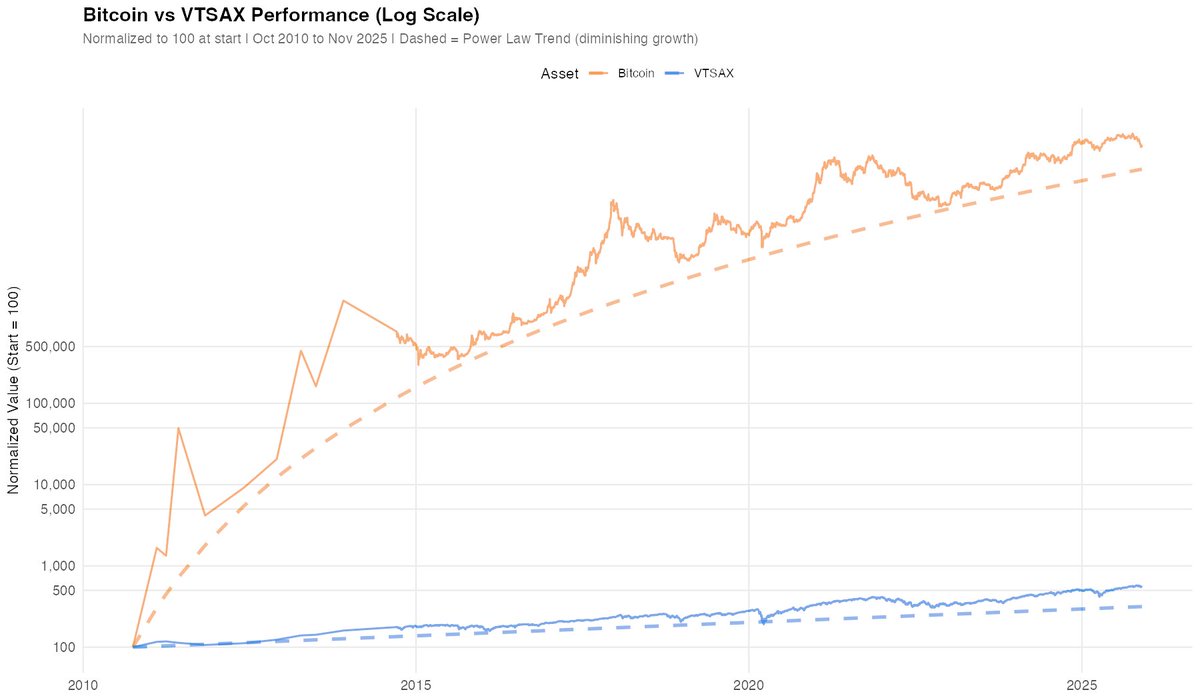
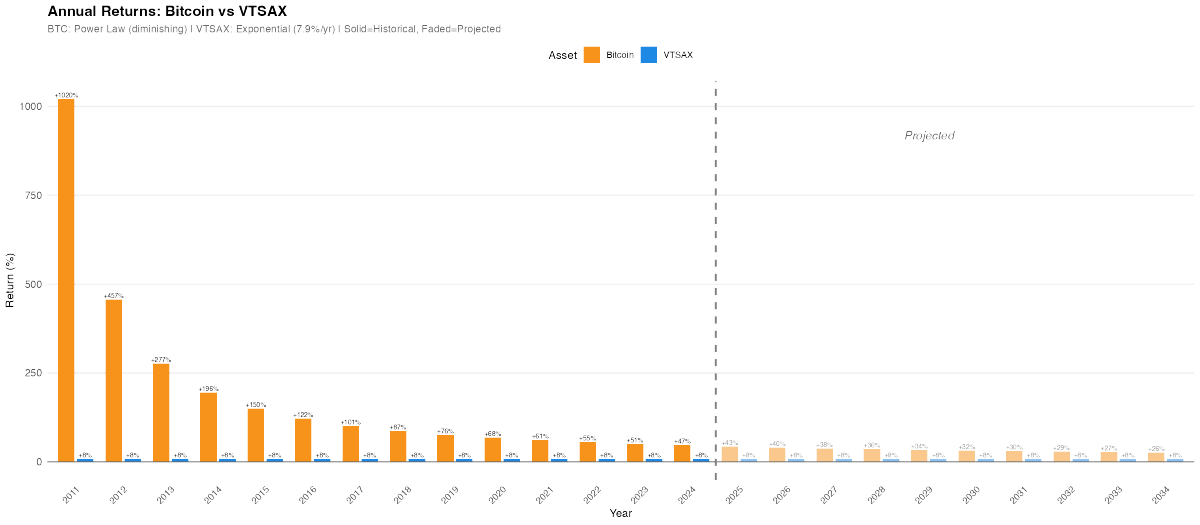
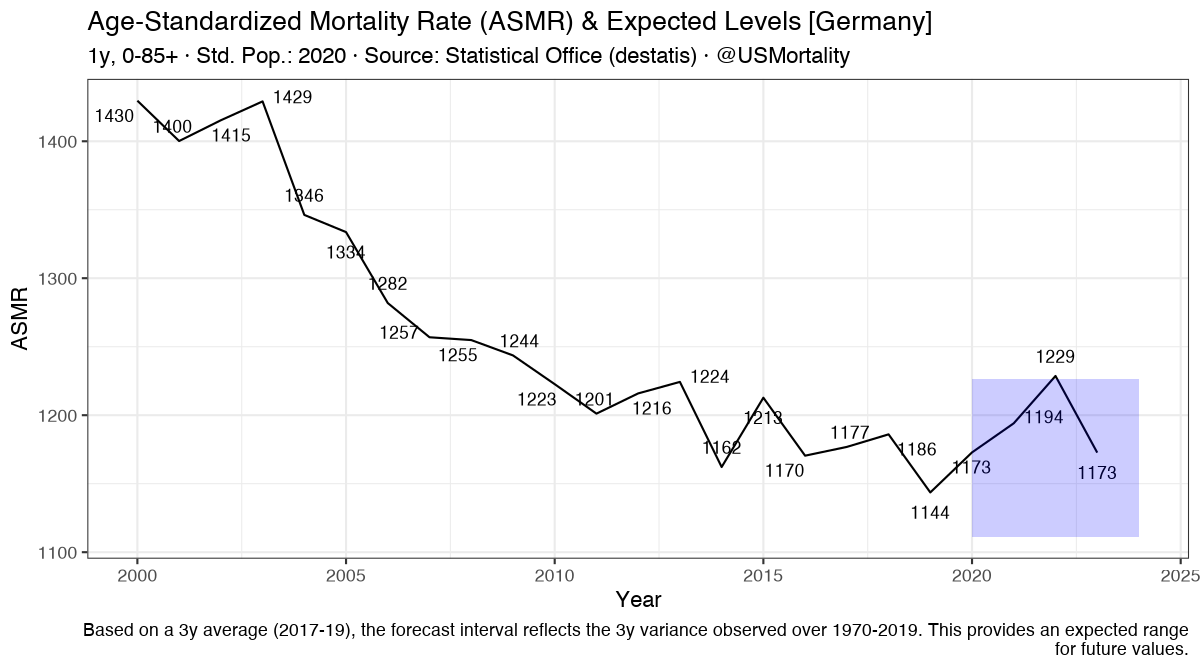
So Florida is apparently getting hit quite hard.
We can see even in Seniors massive excess death.
So I looked at vaccine data...
1/n
We can see even in Seniors massive excess death.
So I looked at vaccine data...
1/n

According to this, we can see that ~80% of Senior Floridians are vaccinated at least once.
Ok, that'd still leave a lot of people, likely hundreds of thousands, unprotected.
2/n
Ok, that'd still leave a lot of people, likely hundreds of thousands, unprotected.
2/n

But then I looked NYT Vaccine Tracker, and found this county, which has extraordinarily low vaccine uptake.
2/n
nytimes.com/interactive/20…
2/n
nytimes.com/interactive/20…

So it must be that there's a lot of covid cases & deaths right?
Nope, there's exactly 0 deaths and 7 cases.
Nope, there's exactly 0 deaths and 7 cases.

Meanwhile, highly vaxxed Miami-Dade, is seeing quite a lot of deaths... (they actually changed reporting here)
I've zipped through other counties... It appears the high-vaxx counties are not doing much better than before...
Is this a general trend? It'd be strange!
I've zipped through other counties... It appears the high-vaxx counties are not doing much better than before...
Is this a general trend? It'd be strange!

• • •
Missing some Tweet in this thread? You can try to
force a refresh