End-to-end learning of multiple sequence alignments with differentiable Smith-Waterman
biorxiv.org/content/10.110…
A fun collaboration with Samantha Petti, Nicholas Bhattacharya, @proteinrosh, @JustasDauparas, @countablyfinite, @keitokiddo, @srush_nlp & @pkoo562 (1/8)
biorxiv.org/content/10.110…
A fun collaboration with Samantha Petti, Nicholas Bhattacharya, @proteinrosh, @JustasDauparas, @countablyfinite, @keitokiddo, @srush_nlp & @pkoo562 (1/8)
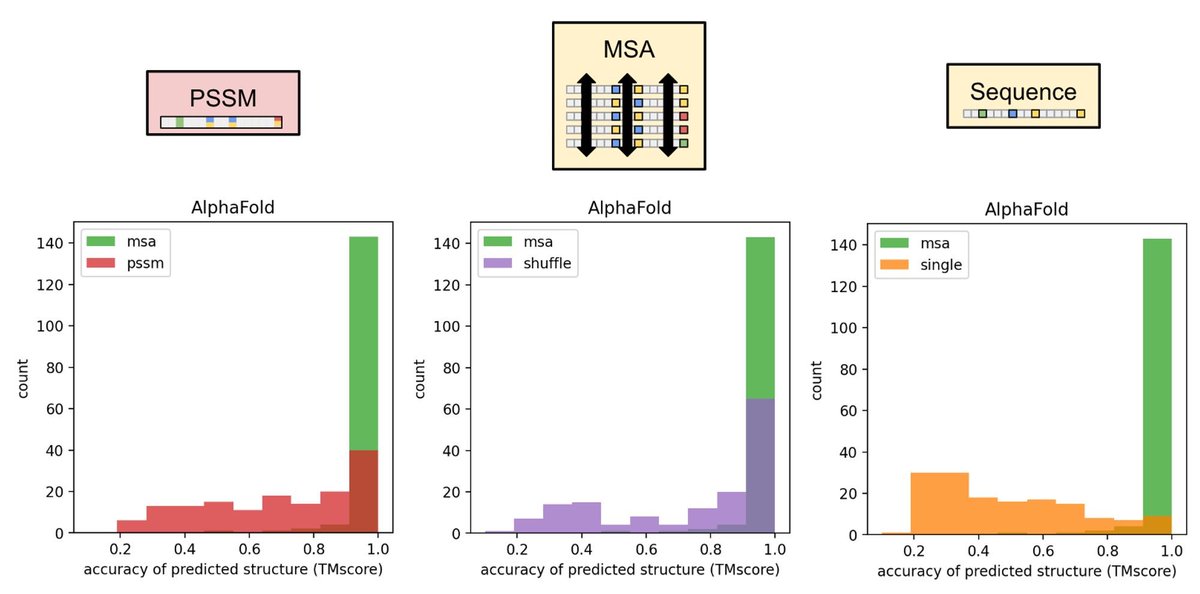
Many methods like GREMLIN, MSA_transformer, RosetTTAFold and AlphaFold rely on input MSA generated by non-differentiable methods. (2/8) 

We ask the question, what if we make the red arrow differentiable and optimize end-to-end. (3/8)

To accomplish this, we implement a differentiable alignment module (LAM). More specifically a vectorized/ striped smith-waterman via #JAX that is extremely fast (4/8)
Special thanks to @jakevdp for #JAX help! 😎
github.com/google/jax/dis…
Special thanks to @jakevdp for #JAX help! 😎
github.com/google/jax/dis…

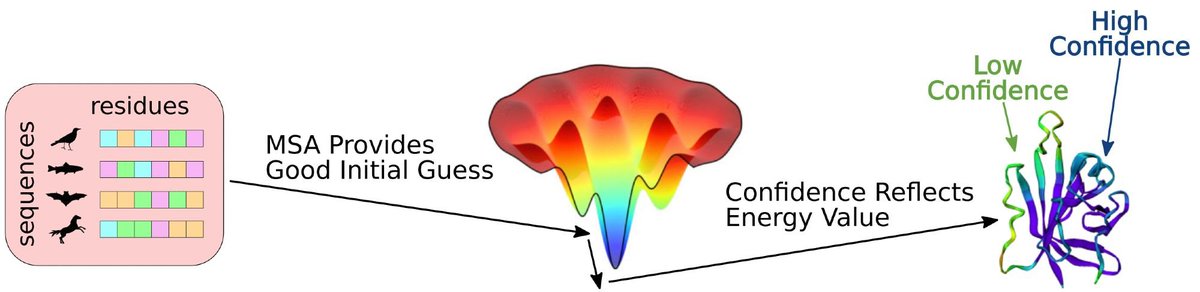
Given AlphaFold and LAM are conveniently implemented in #JAX, as a proof-of-concept, we backprop through AlphaFold and LAM to maximize the confidence metrics (pLDDT and pAE) (5/8)

Maximizing pLDDT (and potentially "learning" a more optimal MSA) often increases structure prediction over our initial input MSAs. (6/8)


LAM also allows us to convert GREMLIN into SMURF (Smooth Markov Unaligned Random Field) that simultaneously learns an MSA, coevolution and conservation of a given rna/protein family. (7/8)

Learning the MSA+Coevolution end-to-end matches and sometimes exceeds the performance of precomputed MSA on proteins and RNA for task of contact prediction. (8/8)

We'll make the code public in a day or two. The owner of our shared GitHub account is currently traveling. 😂
The source code is now public: 👀
github.com/spetti/SMURF
github.com/spetti/SMURF
@jakevdp Note: We are not the first to implement a differentiable alignment module in bio.
Previous Implementations:
bmcbioinformatics.biomedcentral.com/articles/10.11…
Most recent iterations:
pytorch-struct:
github.com/harvardnlp/pyt…
deepblast (cuda)
biorxiv.org/content/10.110…
julia:
live.juliacon.org/talk/QB8EC8
Previous Implementations:
bmcbioinformatics.biomedcentral.com/articles/10.11…
Most recent iterations:
pytorch-struct:
github.com/harvardnlp/pyt…
deepblast (cuda)
biorxiv.org/content/10.110…
julia:
live.juliacon.org/talk/QB8EC8
@jakevdp Oops! Thanks to @thesteinegger for pointing out we had actually implemented an "anti-diagonal" not a "striped" vectorization of smith-waterman.
First described by Wozniak et al. Using video-oriented instructions to speed up sequence comparison. (1997)
bmcbioinformatics.biomedcentral.com/articles/10.11…
First described by Wozniak et al. Using video-oriented instructions to speed up sequence comparison. (1997)
bmcbioinformatics.biomedcentral.com/articles/10.11…

• • •
Missing some Tweet in this thread? You can try to
force a refresh