
Scientist, Assistant Professor @MITBiology, #FirstGen, ProteinBERTologist, 🇺🇦
No Human is illegal.
Moving to: https://t.co/sow6IRD3jj
 Traditional methods approx this signal by taking a multiple sequence alignment of a protein family and computing the inverse covariance matrix. For pLMs we extract it by computing a jacobian over the sequence track (for esm3, structure is masked). (2/8)
Traditional methods approx this signal by taking a multiple sequence alignment of a protein family and computing the inverse covariance matrix. For pLMs we extract it by computing a jacobian over the sequence track (for esm3, structure is masked). (2/8)
 @BrianHie @pdhsu No strong long-range contacts though... (2/3)
@BrianHie @pdhsu No strong long-range contacts though... (2/3) 
 We believe AlphaFold has learned some approximation of an "energy function" and a limited ability to explore. But this is often not enough to find the correct conformation, and often an MSA is required to reduce the search space. (2/9)
We believe AlphaFold has learned some approximation of an "energy function" and a limited ability to explore. But this is often not enough to find the correct conformation, and often an MSA is required to reduce the search space. (2/9) 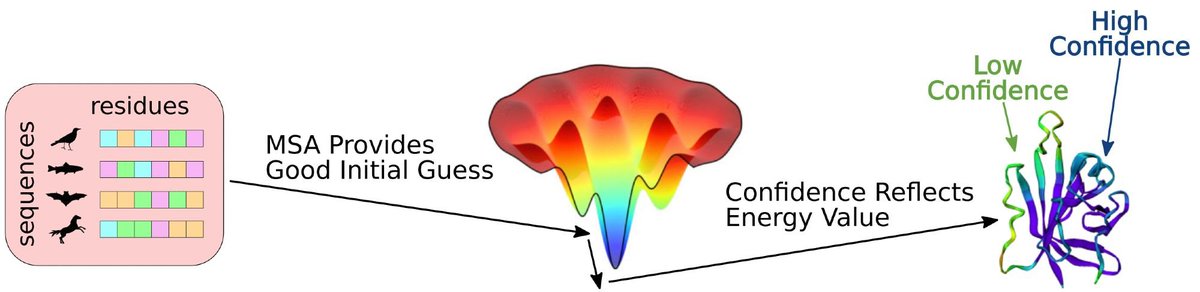
 What do you think will happen if this offset matrix is used instead? [answer will be posted later] (2/3)
What do you think will happen if this offset matrix is used instead? [answer will be posted later] (2/3) 





 @Alexis_Verger
@Alexis_Verger 
 In the spirit of hacking methods for what they were not trained to do. I added support for chain breaks using the residue index offset trick! 😇
In the spirit of hacking methods for what they were not trained to do. I added support for chain breaks using the residue index offset trick! 😇 
 Here is the Pseudo-code of the changes we made and the effects on compile time. (2/2)
Here is the Pseudo-code of the changes we made and the effects on compile time. (2/2) 
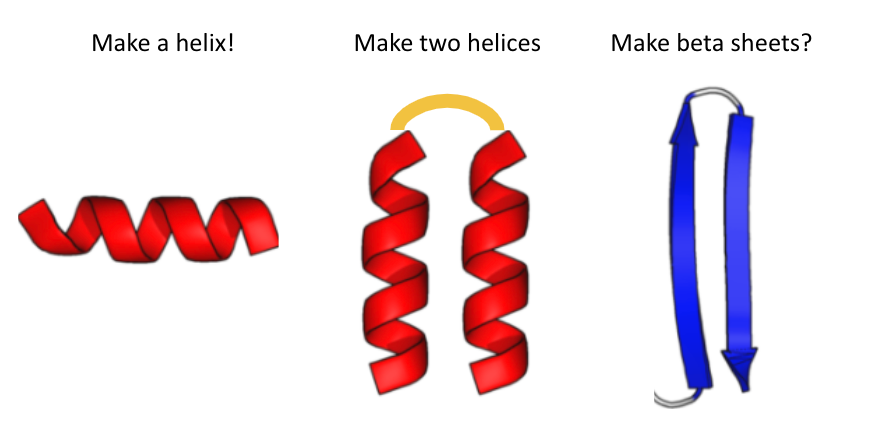 I gave them the following cheat sheet: 😅 (2/5)
I gave them the following cheat sheet: 😅 (2/5) 
