ALERT
Is WIV Sloppy with BL4 viruses?
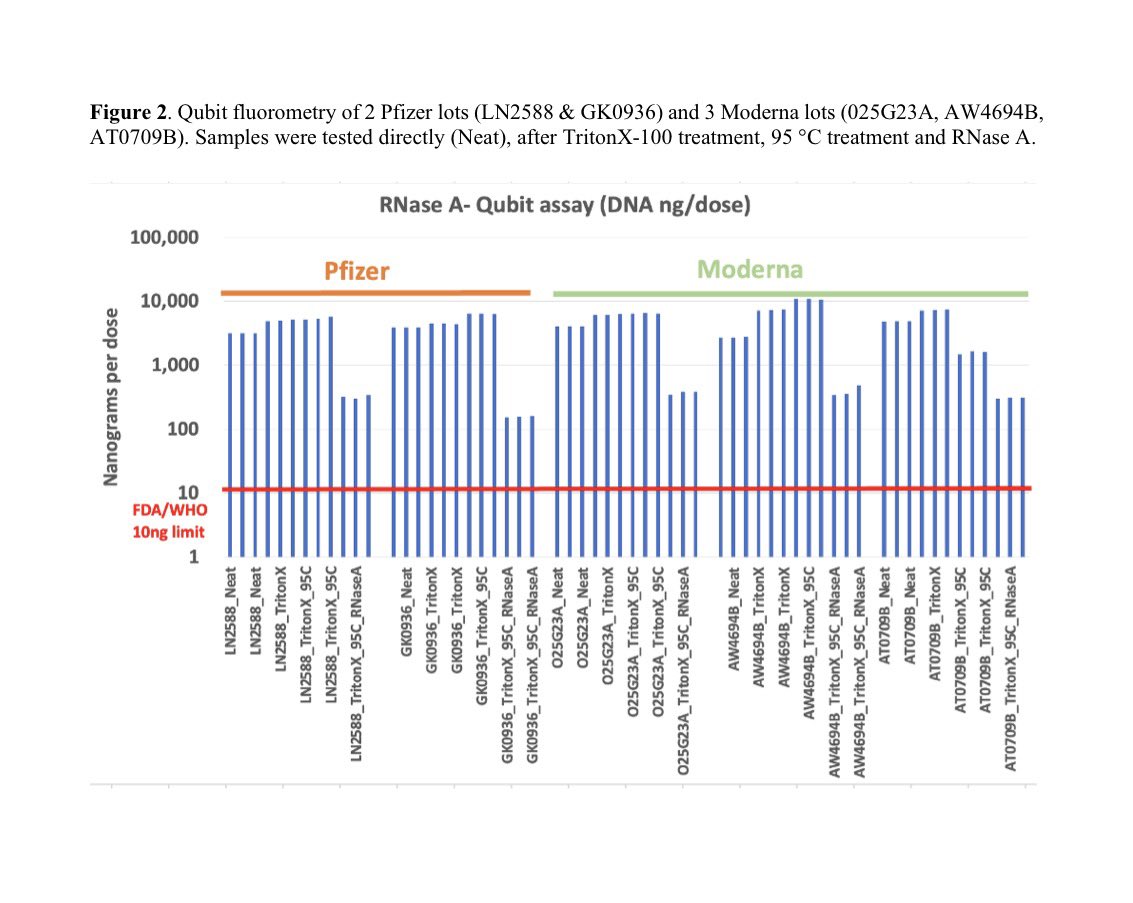
What is Nipahvirus sequence doing in C19 patients sequenced by Wuhan Institute of Virology (WIV)?
This is a BL4 virus far more deadly than C19. Why is it found in patients WIV sequenced and put into NCBI?
Did they really just leak a BL4?
Is WIV Sloppy with BL4 viruses?
What is Nipahvirus sequence doing in C19 patients sequenced by Wuhan Institute of Virology (WIV)?
This is a BL4 virus far more deadly than C19. Why is it found in patients WIV sequenced and put into NCBI?
Did they really just leak a BL4?

First you need to download the Reads referenced in the paper. PRJNA605983. This is an NCBI accession number for a BioProject that will lead you to many SRR#s related to the samples within the BioProject.

You will need 4 open source software tools that run in Linux/Ubuntu.
SRAtoolkit -Downloads reads from NCBI
samtools- general file I/O
Trim_galore- Trims reads of adaptors
bwa mem- Maps reads to a reference.
SRAtoolkit -Downloads reads from NCBI
samtools- general file I/O
Trim_galore- Trims reads of adaptors
bwa mem- Maps reads to a reference.
You will need lots of disk space.
Once you have SRAtoolkit installed, you can use fastq-dump.
kevinmc$ fastq-dump --accession SRR11092057
you will get 2 fastq files (16Gb each). You can us --gzip to download compressed and the next steps can work with zipped files.
Once you have SRAtoolkit installed, you can use fastq-dump.
kevinmc$ fastq-dump --accession SRR11092057
you will get 2 fastq files (16Gb each). You can us --gzip to download compressed and the next steps can work with zipped files.
Next step is to Trim the Illumina adapters off the sequence. Every Target DNA molecule has primer sequences adapted to them for PCR and Sequencing. Need to remove them before alignment to a ref.
/NGS/tools/TrimGalore/trim_galore --paired SRR11092059_1.fastq SRR11092059_2.fastq
/NGS/tools/TrimGalore/trim_galore --paired SRR11092059_1.fastq SRR11092059_2.fastq

This leaves you with some large files and a few report files itemizing how aggressively it trimmed the reads.

Final step is to map these reads against Nipah Virus.
You will need to download this genome accession number.
The Quay paper is good for this. They reproduced the original short preprint paper. I wanted to reproduce this with different tools to ensure it's legit.
AY988601.1
You will need to download this genome accession number.
The Quay paper is good for this. They reproduced the original short preprint paper. I wanted to reproduce this with different tools to ensure it's legit.
AY988601.1
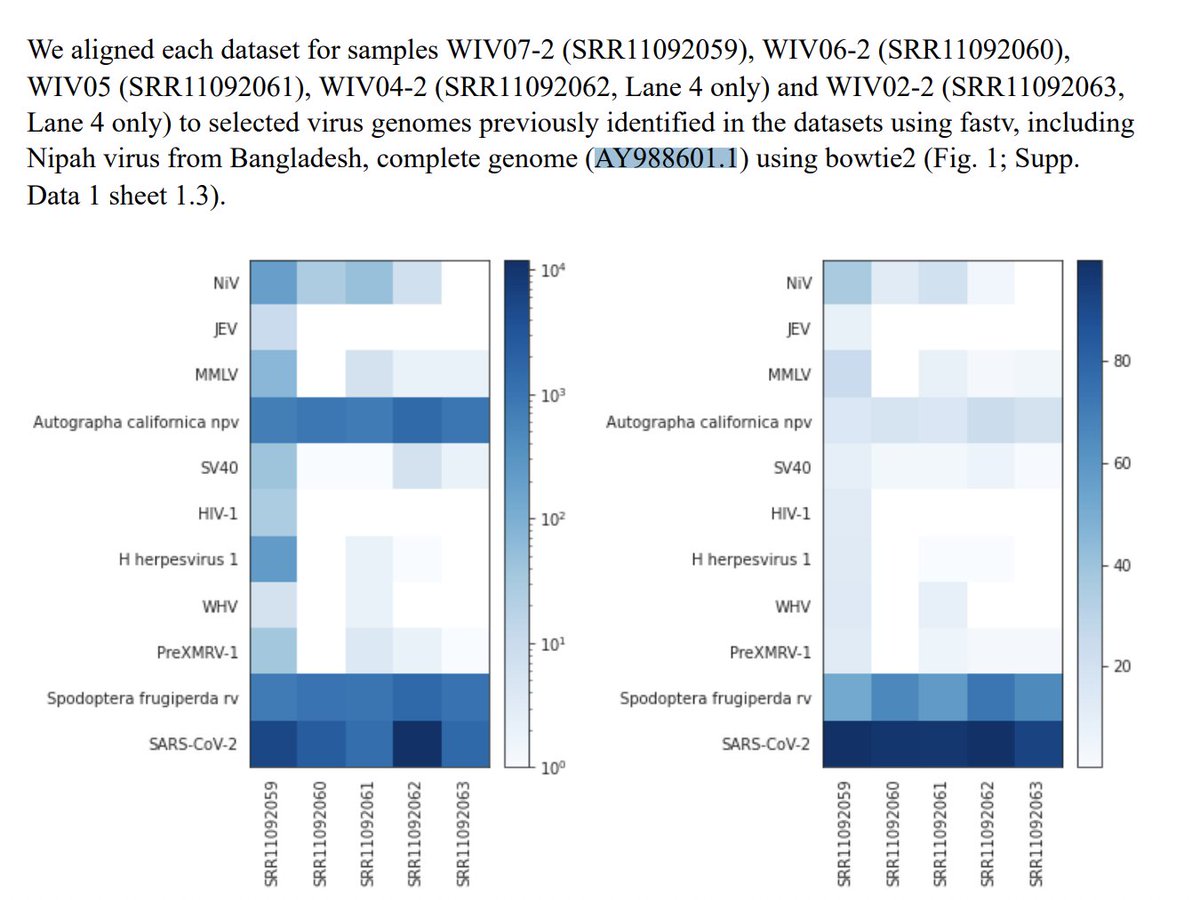
The Quay paper is good read. They went deeper on this problem and found more smoke.
arxiv.org/pdf/2109.09112…
arxiv.org/pdf/2109.09112…
Time to Map the reads with BWA MEM.
-t 4 is the threads used. 1 reference genome, 2 read files and and output piped into samtools to make a bam file.
bwa mem -t 4 ../Nipah_AY988601.1.fasta SRR11092059_1_val_1.fq SRR11092059_2_val_2.fq | samtools sort -o AY_WIV_x_Nipah.bam -
-t 4 is the threads used. 1 reference genome, 2 read files and and output piped into samtools to make a bam file.
bwa mem -t 4 ../Nipah_AY988601.1.fasta SRR11092059_1_val_1.fq SRR11092059_2_val_2.fq | samtools sort -o AY_WIV_x_Nipah.bam -
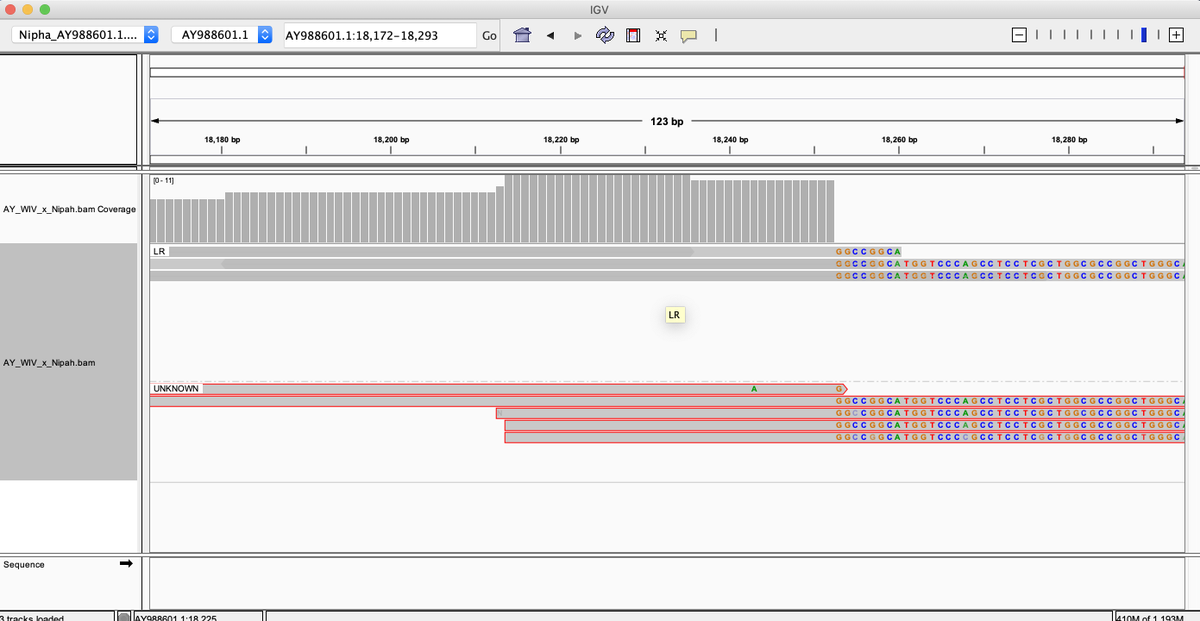
What do we see?
If you want to visualize a BAM file you will need to index the file and use IGV to open it.
samtools index AY_WIV_x_Nipah.bam
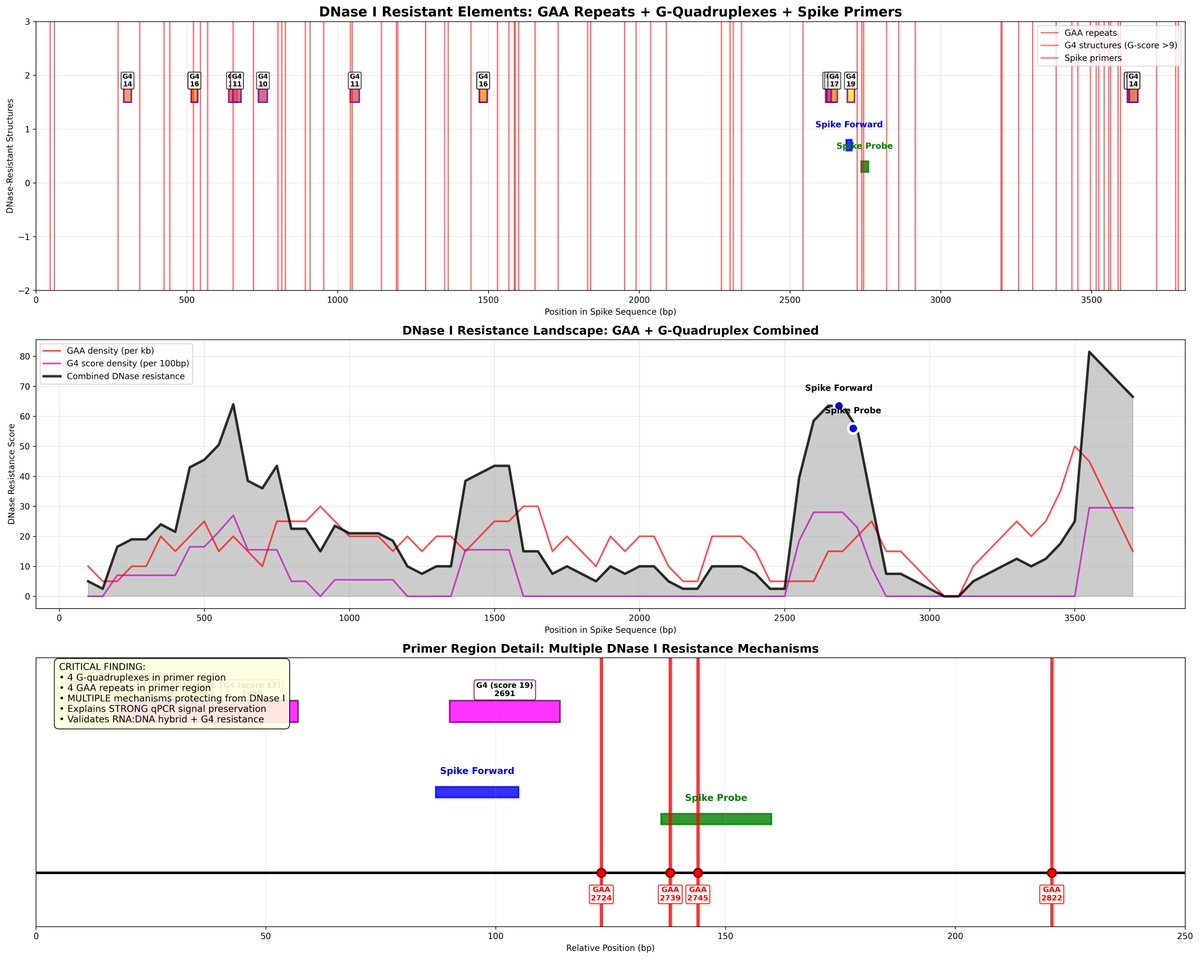
Not only are reads found that map to many parts of the Nipah virus, the reads that map to the end of the virus have novel sequence.
If you want to visualize a BAM file you will need to index the file and use IGV to open it.
samtools index AY_WIV_x_Nipah.bam
Not only are reads found that map to many parts of the Nipah virus, the reads that map to the end of the virus have novel sequence.
The read mapping profile across the genome reproduces Quay et al.
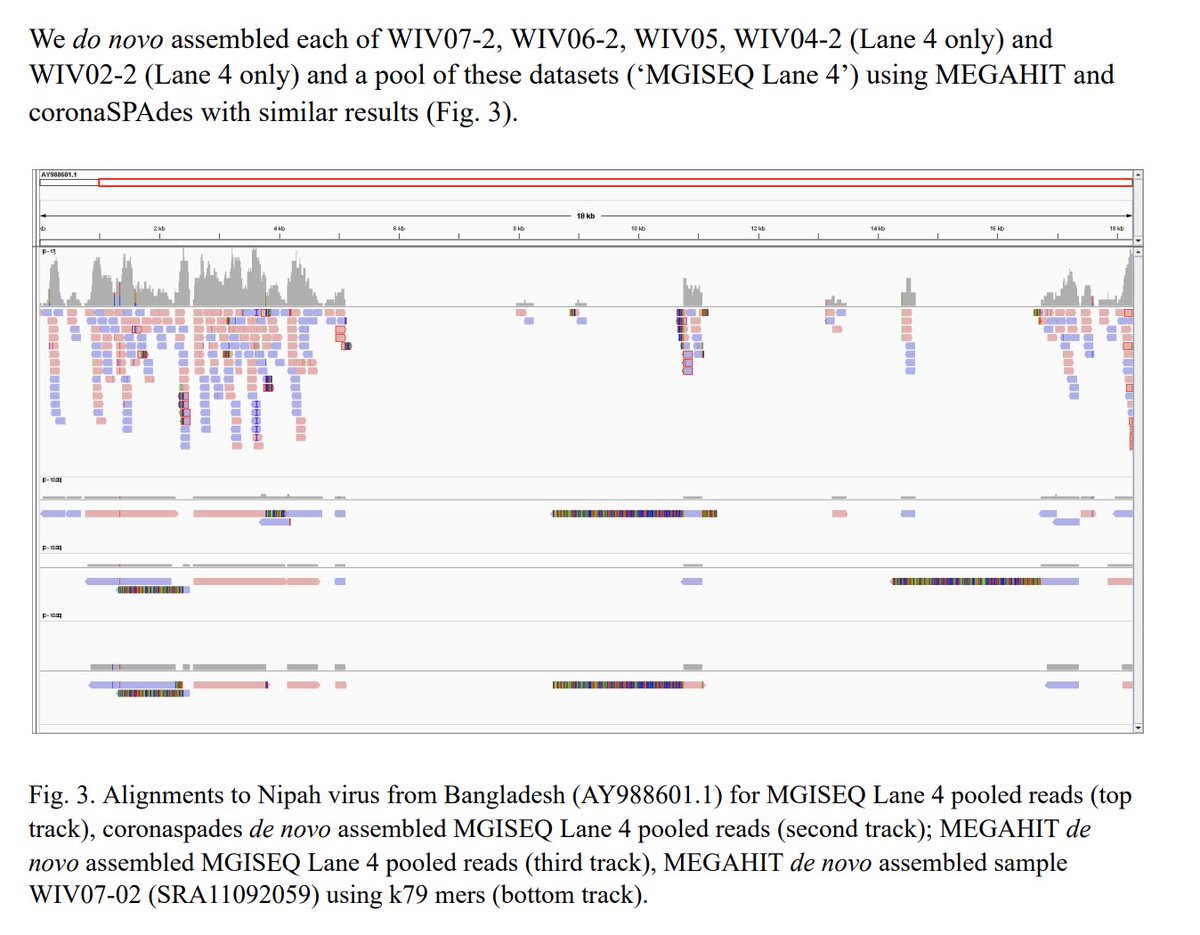
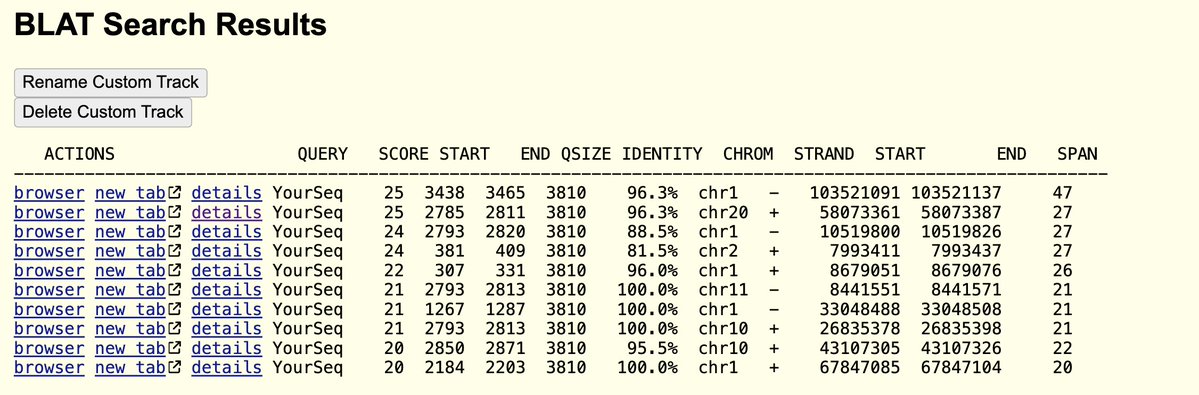
Quay et al, noticed the reads that map to the ends of the virus had novel sequence on them that matched a cloning vector. This is evidence WIV is probably performing GOF on Nipah virus and that the patient may not have been infected by Nipah but that the lab is dirty.
Here is an example of reads that reach into the cloning vector at the end of the virus. The lower reads boxed in red are paired end reads where the reverse read doesn't land on the reference. A BLAST of these unanchored paired reads hits deep into the cloning vector. Makes sense.
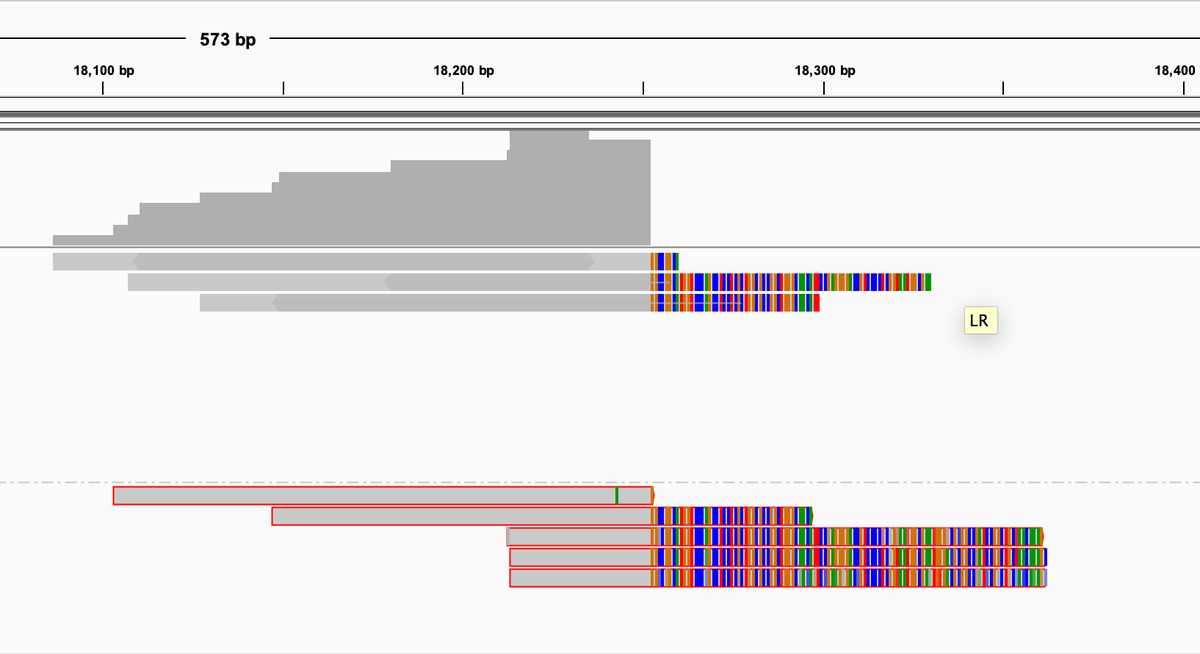
Internal regions of the virus also have regions like this which is odd. Need to dig more here.
This is solid Evidence that WIV is gambling with even more deadly viruses. They need to explain this mess. The people that funded them to do this have a lot to answer for.
Who funds this stuff?
The usual villains.
US tax payers.
news.cornell.edu/stories/2020/1…
Who funds this stuff?
The usual villains.
US tax payers.
news.cornell.edu/stories/2020/1…
The story wouldn't be complete without NIH grants to EcoHealth and Peter Daszak involvement.
The people making the superbugs have royalties for the jab (2 Proline Patent from NIH licensed to both Moderna and Pfizer).
This needs to end.
@RandPaul
npr.org/sections/goats…
The people making the superbugs have royalties for the jab (2 Proline Patent from NIH licensed to both Moderna and Pfizer).
This needs to end.
@RandPaul
npr.org/sections/goats…


The take home message. WIV is handling BL4 viruses and cant seem to keep them from getting into BL2 sequencing projects. Its a mess.
Fauci is funding this work by laundering the money through a fall guy. DARPA rejected funding this work as it was too dangerous. Doesnt stop Fauci
Fauci is funding this work by laundering the money through a fall guy. DARPA rejected funding this work as it was too dangerous. Doesnt stop Fauci
He just finds a few loop holes through the regs he authored and sends it to a sweat shop in China that can't keep their chocolate from getting in their peanut butter.
It probably helps that his wife is on the Ethics committee.
He needs to removed and investigated.
It probably helps that his wife is on the Ethics committee.
He needs to removed and investigated.

.@Daoyu15 @w_mccairn @Harvard2H
Old news to many of you but I checked over the reads and agree with your work.
Old news to many of you but I checked over the reads and agree with your work.
I’m not advocating any violence or Chinaphobia.
Fauci has his hands all over this.
We need to clean up our own house as opposed to marinating in jingoism.
Fauci has his hands all over this.
We need to clean up our own house as opposed to marinating in jingoism.
• • •
Missing some Tweet in this thread? You can try to
force a refresh