
Evidence for ‘live’ RaTG13 at WIV, Part 2
In this thread I dissect the microbial taxa present in the RaTG13 dataset and show they are inconsistent with a fecal swab sample
In this thread I dissect the microbial taxa present in the RaTG13 dataset and show they are inconsistent with a fecal swab sample
2/ Using Metaxa2, only 1.8 % of the reads in the RaTG13 dataset (GSA CRR122287) correspond to small subunit rRNA sequences. This contrasts with 20.7 % present in a Rhinolophus sp. anal swab sample from the WIV (NCBI MN611522) 

3/ This implies the RaTG13 sample underwent rRNA depletion, in contrast to the anal swab sample. This is an optional step when using the TruSeq library preparation kit, indicated as being used on the RaTG13 GSA webpage

4/ The ratio of eukaryotic to bacterial rRNAs is 8.3:1 in the RaTG13 sample, but 1:5.4 in the anal swab sample. The domination of eukaryotic sequences is consistent with a MG-RAST analysis by @MonaRahalkar @BahulikarRahul who compared the same two datasets
hwww.preprints.org/manuscript/202…
hwww.preprints.org/manuscript/202…
5/ This high proportion of eukaryotic rRNAs is also consistent with the observation that 87.5 % of reads map to the Rhinolophus ferrumequinum genome, described in Thread #1
https://twitter.com/stevenemassey/status/1460592468697747466?s=20
6/ The high ratio of eukaryotic:bacterial rRNA sequences in the RaTG13 data is inconsistent with a fecal sample, which should be dominated by bacteria, as found by other investigators. I have built my analysis on their pioneering work:
preprints.org/manuscript/202…
preprints.org/manuscript/202…
7/ Contd:
preprints.org/manuscript/202…
onlinelibrary.wiley.com/doi/10.1002/bi…
drasticresearch.org/2021/04/01/2-i…
zenodo.cern.ch/record/3987503…
vixra.org/abs/2010.0164
zenodo.org/record/4477081…
preprints.org/manuscript/202…
onlinelibrary.wiley.com/doi/10.1002/bi…
drasticresearch.org/2021/04/01/2-i…
zenodo.cern.ch/record/3987503…
vixra.org/abs/2010.0164
zenodo.org/record/4477081…
8/ Bacterial rRNAs provide a fingerprint which can indicate the origin of a sample. Metaxa2 phylogenetic analysis of bacterial rRNA in the RaTG13 data reveals they are dominated by Lactococcus spp. (64 % of bacterial rRNAs)
9/ Lactococcus spp. are lactic acid bacteria and are not typical of the gut. Consistent with this they only comprise 0.07 % of the bacterial rRNAs in the anal swab sample
10/ 4.5 % of RaTG13 bacterial rRNAs belong to Micrococcus spp. Micrococcus spp. are strictly aerobic and are not expected in the gut, which is an anaerobic environment. Consistent with this they comprise 0 % of bacterial rRNAs in the anal swab sample


11/ 18.1 % of RaTG13 bacterial rRNAs belong to Enterobacteriaceae spp. These are typical gut bacteria, but are also common contaminants in cell culture. For example, they dominate a WIV Rhinolophus sinicus transcriptome dataset (SRA SRR5819066) (90.7 % of bacterial rRNAs)

12/ This likely represents a cell culture contamination, consistent with the low diversity of bacterial taxa present, and high ratio of eukaryotic:bacterial rRNAs (88.7:1). So, the presence of Enterobacteriaceae spp. in the RaTG13 data is not itself diagnostic of fecal material
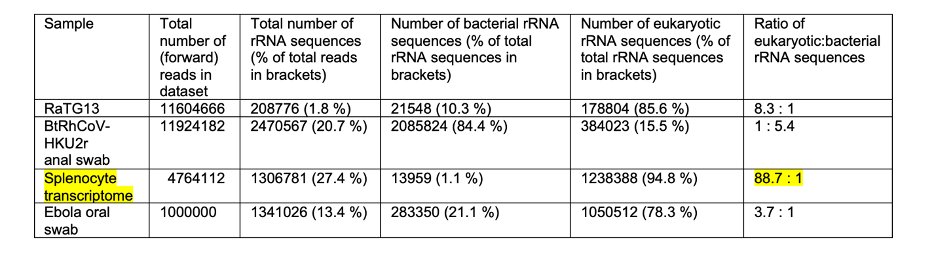
13/ The anal swab rRNA sequences have numerous taxa that are commonly associated with the gut, in contrast with the RaTG13 dataset. I will now systematically go through these

14/ Helicobacter spp. are diagnostic of the mammalian stomach and gut. They comprise 0.4 % of bacterial rRNAs in the anal swab dataset, but only 0.005 % in the RaTG13 dataset
15/ Lachnospiraceae spp. are some of the most abundant members of the gut microbiome in humans. They comprise 6.2 % of bacterial rRNAs in the anal swab dataset, but only 0.7 % of the RaTG13 dataset
16/ Clostridium spp. are a major component of the intestinal tract microbiome. They comprise 47.8 % of the bacterial rRNAs in the anal swab dataset, but only 0.7 % of the RaTG13 dataset
17/ Peptostreptococcaceae spp. are anaerobes found in the human gut, soil and sediments. They comprise 21.2 % of bacterial rRNAs in the anal swab dataset, but only 0.07 % in the RaTG13 dataset
18/ In conclusion, the proportion and identity of bacterial rRNAs present in the RaTG13 data are not consistent with fecal material. This, combined with the mapping results from thread #1 confirm the RaTG13 genome sequence was not generated from a fecal swab
19/ In my next thread I will consider whether RaTG13 was generated from a mislabelled oral swab. Coming soon !
20/ FYA @BillyBostickson, @TheSeeker268, @franciscodeasis, @Daoyu15, @pathogenetics, @mrandersoninneo @Ayjchan @R_H_Ebright @gdemaneuf @MonaRahalkar @Muller_Lab @DaveAtkinsonMD @FrancescoEsp33 @angoffinet @alexandrosM @AntGDuarte @breakfast_dogs @CharlesRixey @humblesci
21/ @Biol4Ever @BlockedVirology @scotub @quay_dr @InWuchang @lab_leak @CapeloMartinez @notoriousFIL @MJnanostretch @SharriMarkson @HansMahncke @ianbirrell @mattwridley @WhiteCoatWaste @sciencecohen @Fynnderella1 @thackerpd @FilippaLentzos @Bobby_Network @dasher8090
• • •
Missing some Tweet in this thread? You can try to
force a refresh



