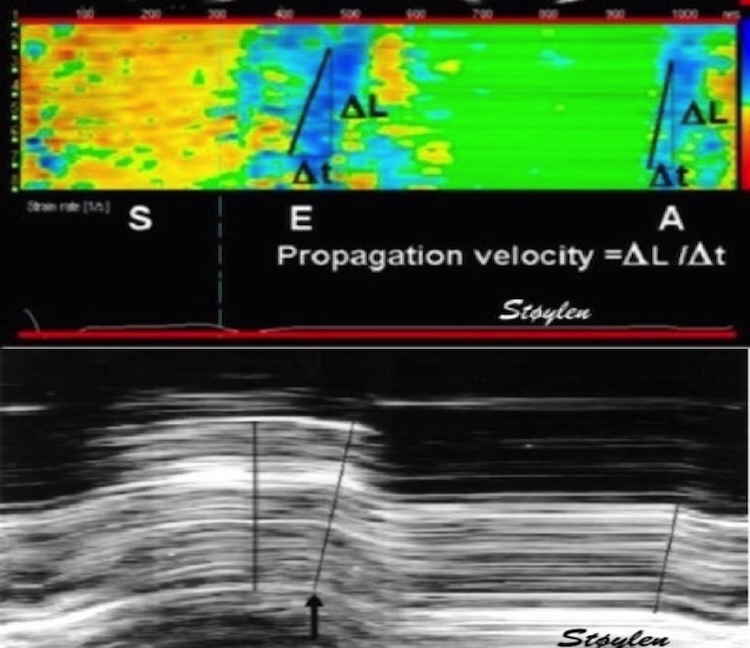
🧵While it is doubtful that GLS adds anything to MAPSE in global function, strain (and strain rate) are useful to assess differences in regional function, both in CHD and dyssynchrony. Regional myocardial work, however, doesn't seem to add information.
1/ The typical finding in CHD is delayed onset and reduced magnitude of systolic shortening, and post systolic shortening in affected segments as seen here in the apex (white curve - compare normal magenta curve). 

2/ In segmental length-pressure loops, using a reconstructed pressure curve, the width of the loop at AVC, corresponds to the strain at AVC. But any regional difference in the loops, must be a function of the difference in strain, as all segments relate to the same pressure curve

In LBBB, the pattern arise from timing differences in the shortening-lengthening cycle betweeseptum and lateral wall, giving shortening of one wall stretching the other, in a complex pattern as seen here.

4/ The "wasted work" arises when one wall shortens, and the other in a passive state is stretched by this shortening, and can be identified by comparing strain from different walls. pubmed.ncbi.nlm.nih.gov/22520537/

5/ Again, the pressure curve is the same for both walls, and the difference in pressure-strain loops must be due only to differences in strain. In this image, the strain curves (below) are plotted against the pressure curve (white) to generate corresponding pressure -strain loops

6/ the "wasted work" seen by clockwise rotations the septal loop, arises from the stretching of the septum simultaneous with pressure rise, but is easily seen by divergence of the primary strain curves. There is no new information.

7/ using the same reconstructed pressure curve, but substituting regional strain with global strain, you get a measure related to global myocardial work. As longitudinal strain is responsible for about 60 - 70% of SV, strain based GMW should be less than SV based GMW.

8/ The area of the PV-loop is the true GMW. The height of the PV-loop is the SBP-LVDBP difference, the width is the SV. Mean SBP and mean LVDBP ((blue dotted rectangle), shows the relation in an easy way. GMW is SV x (mean SBP - mean LVDBP).

9/ As the reconstructed pressure curve onlu assumes diastolic LV pressure, and SV is available by Doppler or B-mode, I'm uncertain what GLS based GMW adds compared to a simple estimator of SV x SBP.
10/ Being the the product of SV and BP, it's a measure of myocardial work (i.e. = energy expenditure), and definitely load dependent, as preload increases SV, while BP is a measure of afterload, this was shown already by Sonnenblick. pubmed.ncbi.nlm.nih.gov/13978233/

11/ Unlike SV, EF, MAPSE or GLS, which ⬆️ with contractility and preload and ⬇️ with afterload, GMW ⬆️ with contractility, preload AND afterload. This means ⬇️ GMW is bad in HF, while ⬆️ GMW is bad in afterload states as HT or AS. So the clinical impact is uncertain.
• • •
Missing some Tweet in this thread? You can try to
force a refresh