
I made a new plot type to visualize how sub-lineages are growing with respect to their parent in a country.
Using @ProjectJupyter, Voila, and Binder I managed to make it interactive, so that everyone can type in their lineages/countries.
Try it out here: mybinder.org/v2/gh/corneliu…
Using @ProjectJupyter, Voila, and Binder I managed to make it interactive, so that everyone can type in their lineages/countries.
Try it out here: mybinder.org/v2/gh/corneliu…
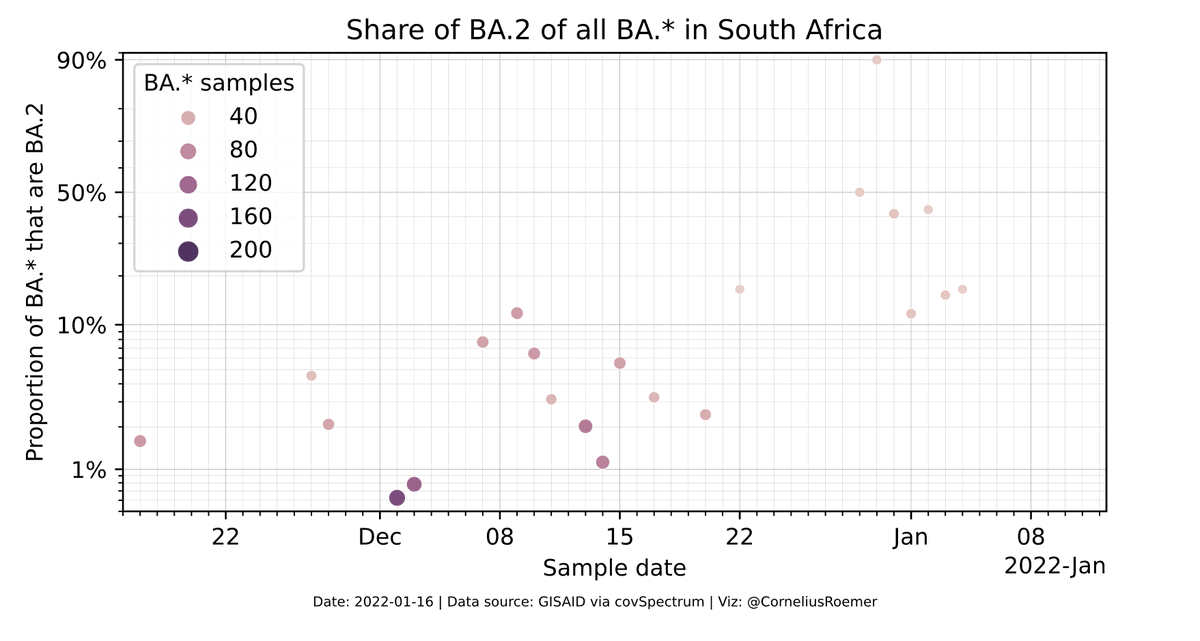
BA.2 is also rising as a share of all Omicrons in South Africa
End of November BA.2 represented around 2% of Omicrons.
Beginning of January, it was about 10 times more, maybe 20%.
End of November BA.2 represented around 2% of Omicrons.
Beginning of January, it was about 10 times more, maybe 20%.
In India, BA.2 has become the dominant Omicron variant towards end of December. Growing from ~5% in mid December to >50% by beginning of January.

In most other countries, BA.1 remains dominant, though BA.2 seems to slowly grow in share everywhere.
Apart from some predictions based on BA.2 sequence by @jbloom_lab not much is known about BA.2.
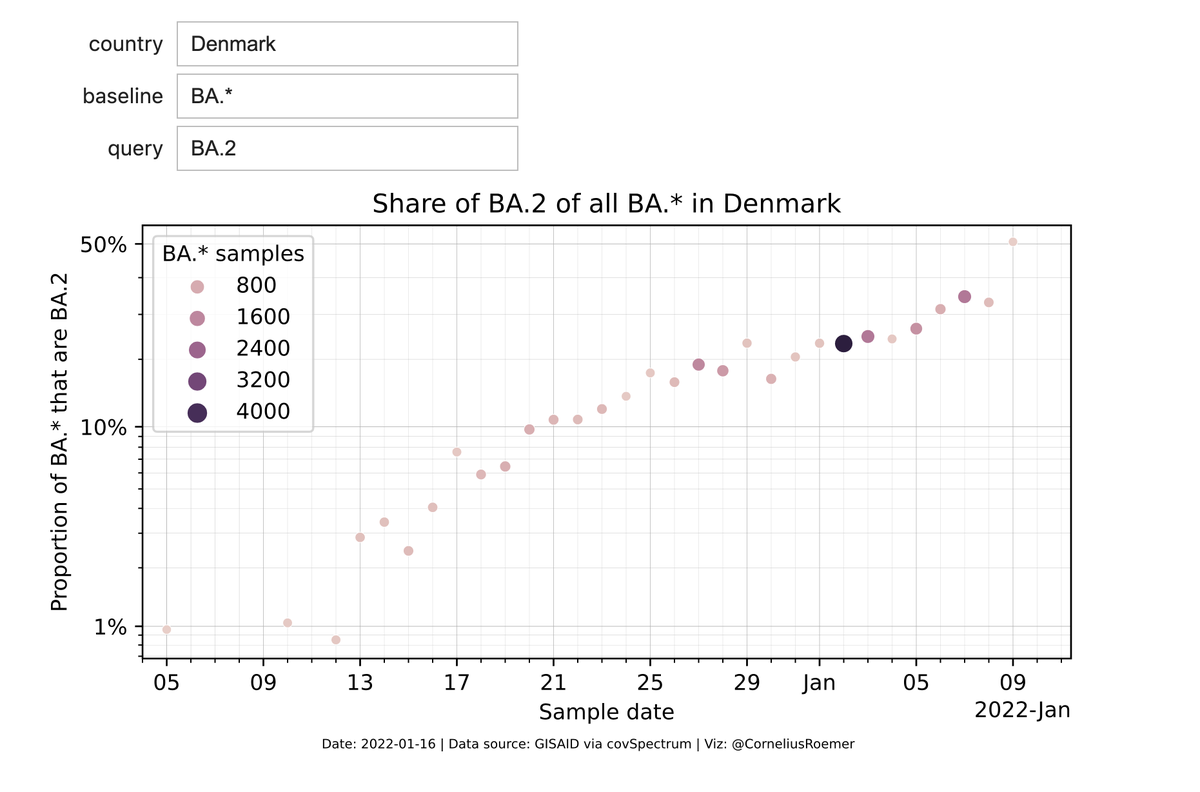
First clinical data will possibly come from Denmark.
Apart from some predictions based on BA.2 sequence by @jbloom_lab not much is known about BA.2.
First clinical data will possibly come from Denmark.
https://twitter.com/jbloom_lab/status/1470442863108362240?s=20

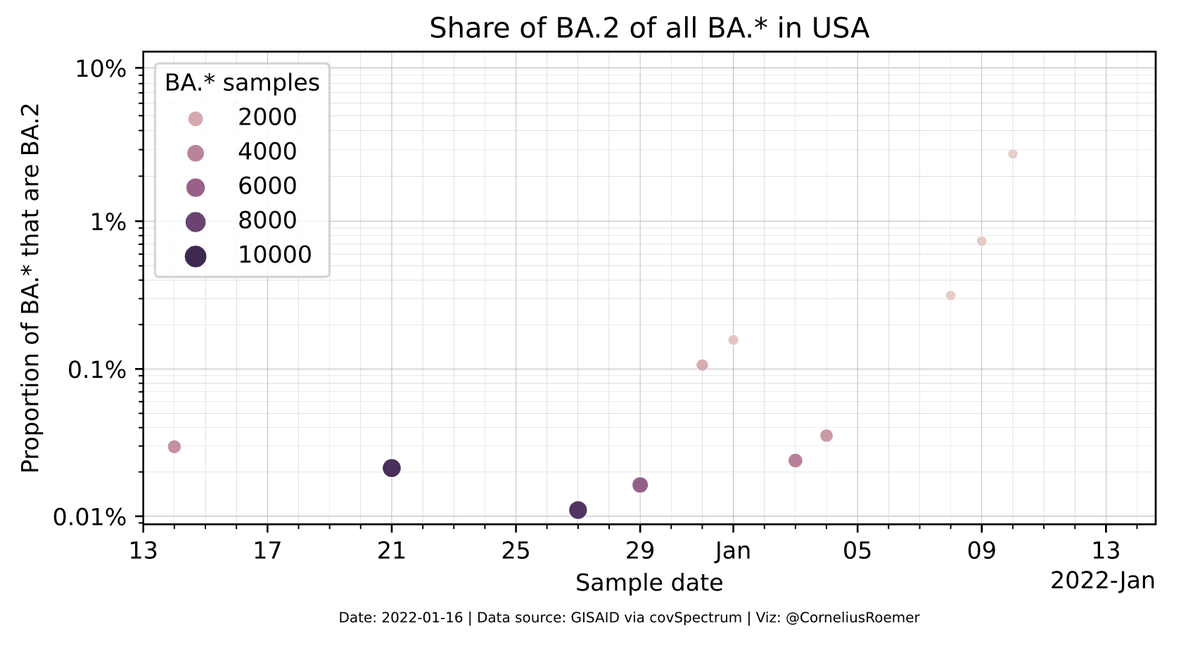
Here's the BA.2/Omicron share in the US
Where there are no dots visible, the share is 0% (which is minus infinity on a logit scale and hence not shown).
Where there are no dots visible, the share is 0% (which is minus infinity on a logit scale and hence not shown).
BA.2 _is_ detectable by PCR, these news reports are totally wrong.
Depending on the PCR test used it may not look like BA.1 (the other Omicron). But it will still give a positive result.
Frustrating to see falsehood about non-detectability still around.
Depending on the PCR test used it may not look like BA.1 (the other Omicron). But it will still give a positive result.
Frustrating to see falsehood about non-detectability still around.
https://twitter.com/i24NEWS_FR/status/1482716087313735683?s=20
The same plot type can be used to compare Delta variants, for example AY.4.2* vs all of Delta.
Here in the UK
Here in the UK

The code is available here: github.com/corneliusroeme…
It's ridiculously easy to set up and deploy.
I had to fight a bit getting the plots to refresh and not error with partial input but now it works splendidly.
Easy to extend, e.g. query for mutations, multi-country etc.
It's ridiculously easy to set up and deploy.
I had to fight a bit getting the plots to refresh and not error with partial input but now it works splendidly.
Easy to extend, e.g. query for mutations, multi-country etc.
• • •
Missing some Tweet in this thread? You can try to
force a refresh







