Compounding error
Transcriptional error and Translational error.
The mRNA jabs use a RNA polymerase to synthesize mRNA from a DNA plasmid.
They replace U with m1pU.
The error rate jumps to 100-300 errors per million bases. That’s 10-^3 -> 10-^4 error
biorxiv.org/content/10.110…
Transcriptional error and Translational error.
The mRNA jabs use a RNA polymerase to synthesize mRNA from a DNA plasmid.
They replace U with m1pU.
The error rate jumps to 100-300 errors per million bases. That’s 10-^3 -> 10-^4 error
biorxiv.org/content/10.110…

The vax is ~4200bp.
This is approximately 1 error in every vax molecule and you get injected with 40T.
This is a result of m1pU which is a low fidelity base. This low fidelity also impacts the next step of tRNA hybridization in translation.
This is compounding error.
This is approximately 1 error in every vax molecule and you get injected with 40T.
This is a result of m1pU which is a low fidelity base. This low fidelity also impacts the next step of tRNA hybridization in translation.
This is compounding error.

Pfizer has mostly sold this base replacement as improving the magnitude and durability of expression.
As with many things in biology, when you optimize for the magnitude of expression, you sacrifice fidelity.
As with many things in biology, when you optimize for the magnitude of expression, you sacrifice fidelity.

Many think of variants as from the virus, not the vax
The virus has an ExoN gene to error correct the polymerase errors. These error correcting proteins don’t like low fidelity bases like m1pU
In order maximize expression of m1pU they need sloppy enzymes
pnas.org/doi/10.1073/pn…
The virus has an ExoN gene to error correct the polymerase errors. These error correcting proteins don’t like low fidelity bases like m1pU
In order maximize expression of m1pU they need sloppy enzymes
pnas.org/doi/10.1073/pn…


As for IVT polymerase error rate, I’d like to emphasize that the folks from New England Biolabs are no hacks when it comes to enzymology.
Their founder Rich Robert’s won the Nobel prize for the discovery of restriction enzymes and they have been the gold standard for decades.
Their founder Rich Robert’s won the Nobel prize for the discovery of restriction enzymes and they have been the gold standard for decades.

The problem is more pronounced with translation.
This model is changing only a few codons. Change them all I bet the error rate escalates.
pnas.org/doi/10.1073/pn…
This model is changing only a few codons. Change them all I bet the error rate escalates.
pnas.org/doi/10.1073/pn…

Add the transcriptional and translational compounded error and you have a combinatorial biochemistry problem on every injection.
This is why there is raw sequence data for vax lots in NCBI.
There is no peptide sequence of one of these mRNA libraries in NCBI.
Just smears on gels.
This is why there is raw sequence data for vax lots in NCBI.
There is no peptide sequence of one of these mRNA libraries in NCBI.
Just smears on gels.

This is a highly variable prodrug.
The final drug has never been characterized.
There are 3D structures of protein translated from mRNA transcribed from a DNA construct without m1pU. Not relevant. No m1pU.
The final drug has never been characterized.
There are 3D structures of protein translated from mRNA transcribed from a DNA construct without m1pU. Not relevant. No m1pU.


There are also smeary antibody stained western blots.
Antibody stains are biased toward error free proteins. They don’t tag the mutated ones.
Need to stain everything.
Cell free in vitro TNT to understand glycosylation or PNGase F for removing in vivo glycosylation. More ?s.
Antibody stains are biased toward error free proteins. They don’t tag the mutated ones.
Need to stain everything.
Cell free in vitro TNT to understand glycosylation or PNGase F for removing in vivo glycosylation. More ?s.


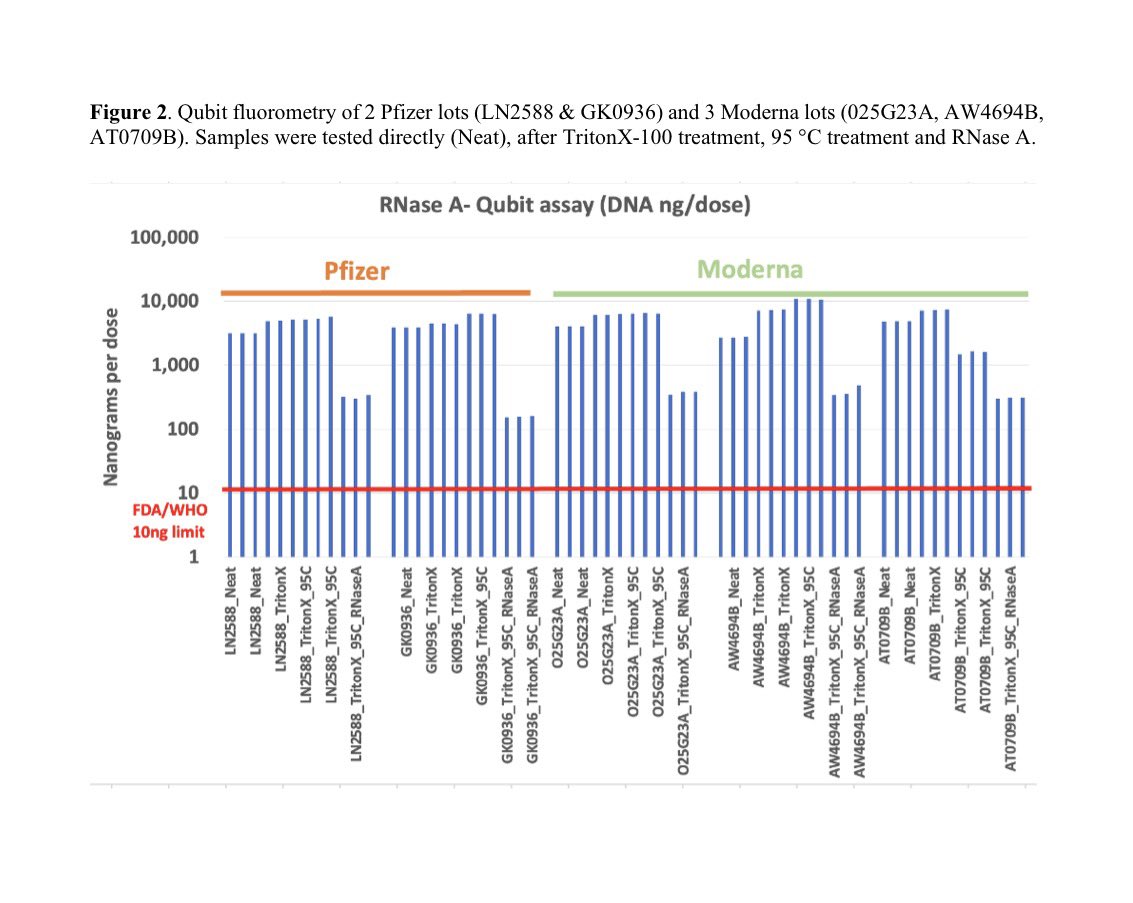
In summary, no mRNA injection should ever escape lot to lot sequence QC that is sensitive enough to find parts per thousand error. 40T molecules injected means small % error = billions of contaminants.
Protein sequencing of the final drug should be required.
Protein sequencing of the final drug should be required.
This is why there is NO lot to lot raw sequence data in NCBI.^^^^
To simplify, the error measured by Chen et al, suggests 1 error in every mRNA molecule. Poisson would imply some molecules have 2 and 3 errors and many have zero.
Now imagine you inject 40 Trillion molecules where each one is different.
The combinatorics are mind blowing.
Now imagine you inject 40 Trillion molecules where each one is different.
The combinatorics are mind blowing.
The good news is that folk at BASE are starting to look at this problem with direct RNA sequencing with Oxford Nanopore (ONT). But the m1pUs look foreign to the ONT platform and get called as both a C and a T. It's unlikely this will have the accuracy to pick up 1:1K heteroplasmy

The bad news is, this should have been done and made public before injecting 1B people. Its pretty bleeding edge so I can see how it was overlooked but at the minimum ILMN sequencing could have measured the heteroplasmies. Probably need UMIs to discount the cDNA syn error.

But now that the camel has its nose in the tent and has feasted on the money machine.. It will be back for more.

For those doubting my assessment of the mfg process, BASE spells it out here. Maybe my purity expectations are too high but I'd be anlot less scrupulous if Pfizer/Moderna put any raw sequence live for lot to lot QC. Zero for the vax. Millions for the virus.

This preprint monitors the translation fidelity in PseudoU and m1PseudoU.
biorxiv.org/content/10.110…
biorxiv.org/content/10.110…



Another one. I dont see an email address for this author.
jpands.org/vol27no2/hatfi…
jpands.org/vol27no2/hatfi…
One reason I don’t like the lemons into lemonade comment in this paper…
It speaks to sloppy translation perhaps being a good defense against variants.
That’s a very sloppy way to achieve a goal and may come with risks.
Consider this paper. RNA-Roulette
biorxiv.org/content/10.110…
It speaks to sloppy translation perhaps being a good defense against variants.
That’s a very sloppy way to achieve a goal and may come with risks.
Consider this paper. RNA-Roulette
biorxiv.org/content/10.110…
If anyone has spare cycles, it would be helpful to calculate the Amino Acid edit distance of The vax Amino Acids to human amino acids in all 6 reading frames. This would give us a framework of the autoimmune risk of these synthesis errors.
The EMA leak has a lot to say about RNA integrity loss during manufacturing scale up.
If I had to guess,
Nucleotide purity probably played a role. It’s hard to scale up the synthesis of modified nucleotides and the polymerases don’t like to incorporate them. They stall.
If I had to guess,
Nucleotide purity probably played a role. It’s hard to scale up the synthesis of modified nucleotides and the polymerases don’t like to incorporate them. They stall.


Particularly at long tracks of U.
PolyU incorporation it pure N1 methyl pseudoU could create polymerase stall points and truncated mRNA.
But RNA integrity numbers also include mRNAs that are too long so one can’t assume it’s only truncated mRNA.
childrenshealthdefense.org/defender/europ…
PolyU incorporation it pure N1 methyl pseudoU could create polymerase stall points and truncated mRNA.
But RNA integrity numbers also include mRNAs that are too long so one can’t assume it’s only truncated mRNA.
childrenshealthdefense.org/defender/europ…
• • •
Missing some Tweet in this thread? You can try to
force a refresh