
10,048,466! That’s a lot of #SARSCoV2 genomes in the single largest phylogeny ever that we update and optimize every single day! Here, I’ll explain how we are doing pandemic-scale phylogenomics. 
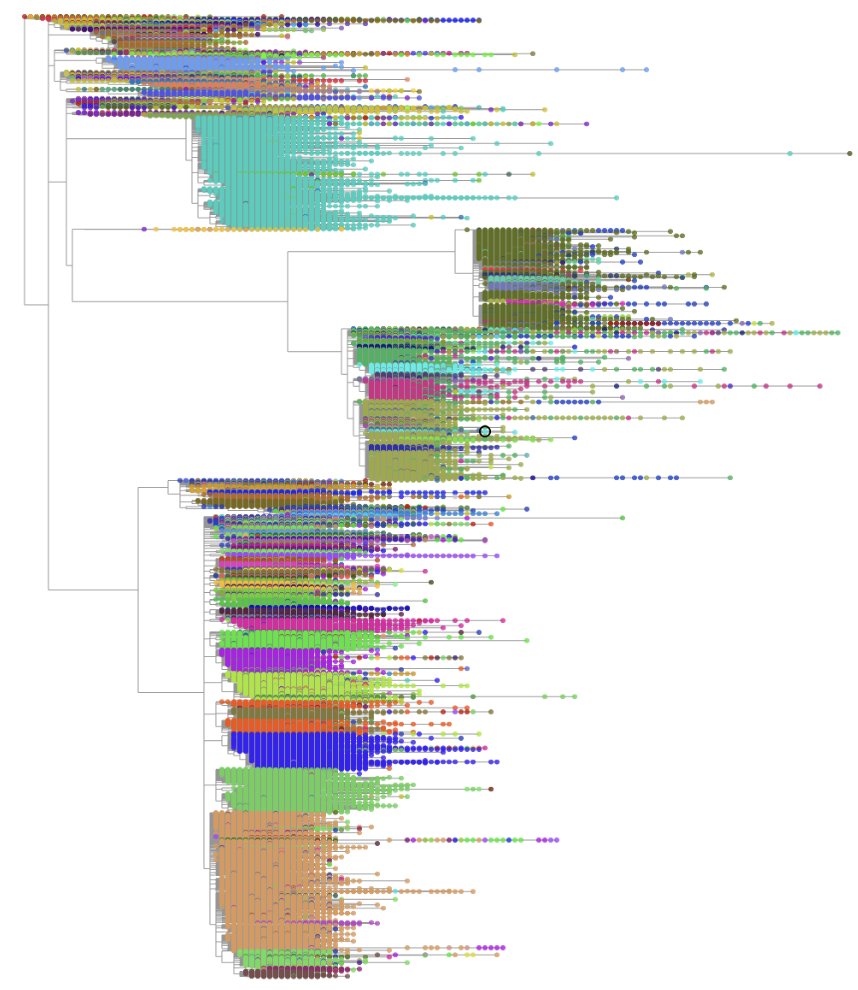
We start by aggregating all of the new SARS-CoV-2 genomes from @GISAID, @NCBI, and @CovidGenomicsUK. After QC, we add each genome to the ever-growing phylogeny using @yatishturakhia’s amazing tool, UShER: nature.com/articles/s4158…
UShER places samples one by one and sometimes it infers suboptimal trees. Thanks to nifty engineering, our tool, matOptimize uses SPR moves to optimize the entire 10M sample tree every day! biorxiv.org/content/10.110…
Amazingly, Bryan Thornlow and @alexkramer_ showed that the trees we infer using parsimony have slightly higher likelihoods than those inferred by many maximum-likelihood programs and ours takes only a tiny fraction of the time. biorxiv.org/content/10.110…
I don’t think that parsimony is inherently better than likelihood. We think the difference is that matOptimize is able to search much more tree space in the same amount of time and that the super dense sampling of SARS-CoV-2 genomes is very well suited to parsimony.
Also check out @Nicola_De_Maio’s amazing “MAximum Parsimonious Likelihood” (MAPLE) method as a way to get the best of both worlds in the near future! biorxiv.org/content/10.110…
Thanks to @AngieSHinrichs, UShER is now a crucial part of genomic epi. It’s the primary basis for new @PangoNetwork lineage designations and assignments, and users worldwide upload their samples to visualize relationships with the other 10M genomes. genome.ucsc.edu/cgi-bin/hgPhyl…
We have had a ton of people contribute! Some of the main folks (in no particular order and certainly non-exhaustive): @AngieSHinrichs, @yatishturakhia, Jakob McBroome, Bryan Thornlow, @alexkramer_, David Haussler, @RobLanfear, @EBIgoldman, @Nicola_De_Maio, and many more.
And of course, sampling all these genomes has been a global effort! Sequence producers all over the world rush to share their data, genomic databases aggregate and distribute these data, and a kajillion open source bioinformatic tools analyze them.
• • •
Missing some Tweet in this thread? You can try to
force a refresh


