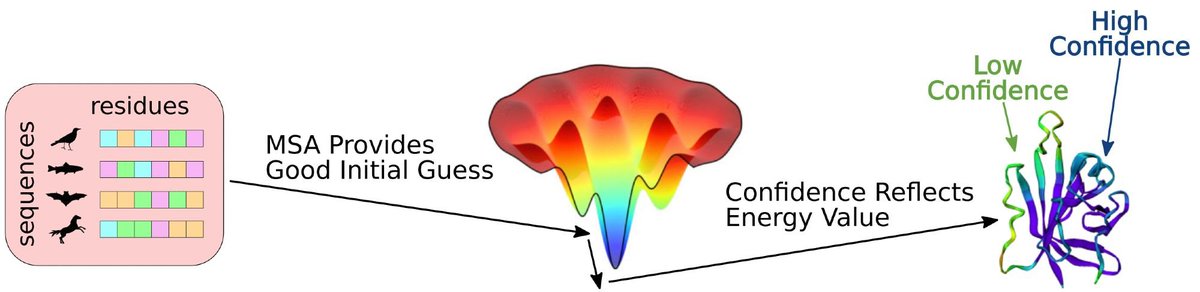
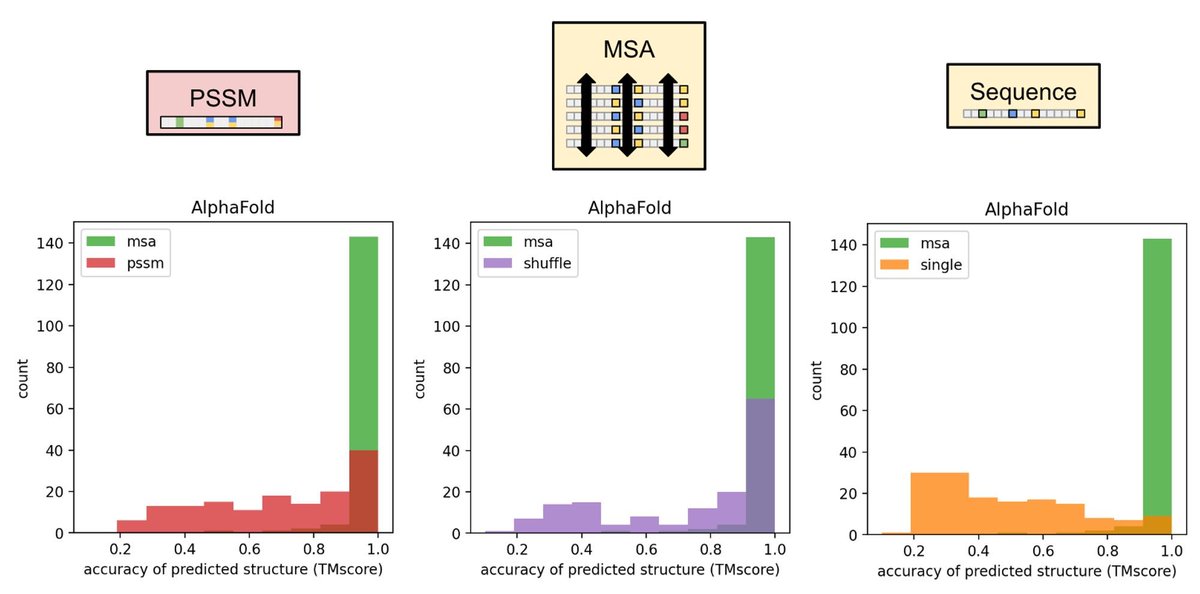
For my latest attempt at introducing proteins to students, I made a Google Colab Notebook that predicts proteins from a single sequence. I asked the students to tweak the sequence to get a helix or two helices or... (1/5)
colab.research.google.com/github/sokrypt…
colab.research.google.com/github/sokrypt…
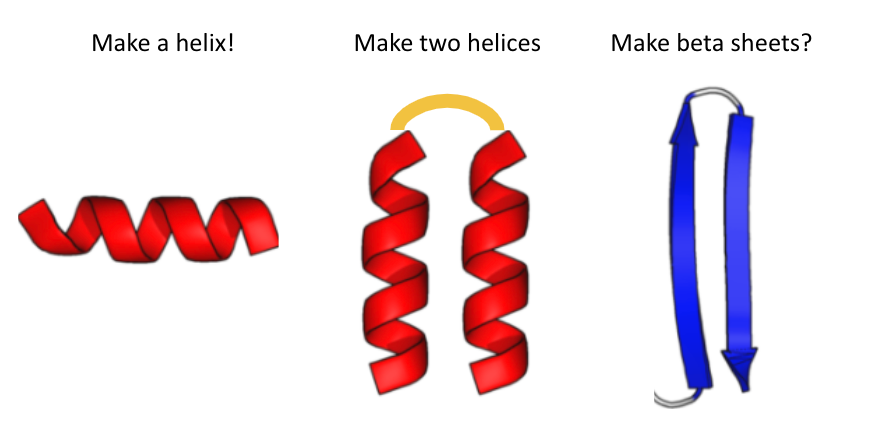
I gave them the following cheat sheet: 😅 (2/5)

To make this practical, I had to make various tweaks to AlphaFold to make it compile as fast as possible (~10 seconds) and run as fast as possible (<1 second) and to avoid recompiling if the sequence length changes or the number of recycles changes. (3/5)
I add @david_koes py3Dmol to allow the display of structure with sidechains colored hydrophobic/hydrophilic (to encourage students to think about hydrophobicity). Finally, I add the ability to animate the proteins through recycles at the end. 🙃 (4/5)

Any feedback is welcome! Share your "designed" proteins 😁 and remember: (5/5)

For those that are curious about how I reduced the compile time. I noticed AlphaFold was using a python for-loop for its fold_iteration (within /folding.py). I replaced this with a jax scan fn, reducing compile time from a few of mins to 20 secs. (1/2)


Finally, I only compile a single pass of the model and move the recycling mechanism outside of the compiled code. This lets me run as many ♻️ as I want externally. I just needed to add an option (to /modules.py) to input/output the previous pair/pos via batch. (2/2)

The advantage of moving recycles outside of the compiled code is it allows you to save all the outputs at each ♻️...(allowing for cool animations) and if you are not happy with the result, you can technically resume ♻️ling... without rerunning prev. iterations 🤓 (3/2)
(for Jax people looking at this code. AlphaFold is using some custom "safe" key splitting... And the scan fn is unable to iterate over the split keys... To get around this issue, I added key splitting within the function. Maybe there is a better solution?)
I figured it out! 😀 @agvaughan

• • •
Missing some Tweet in this thread? You can try to
force a refresh