
@humblesci Daoyu @ydeigin @quay_dr and myself have published a study that shows the exclusion of 20 A/B intermediate SARS2 genomes from the analysis of Pekar et al. is unwarranted 🧵
zenodo.org/record/7005332…
zenodo.org/record/7005332…
2/ The existence of such intermediate genomes in humans is incompatible with the two spillover model for the origin of C19
To recap: lineage A has T8782/C28144 (T/C) while lineage B has C8782/T28144 (C/T)
An A/B intermediate will have C/C or T/T
To recap: lineage A has T8782/C28144 (T/C) while lineage B has C8782/T28144 (C/T)
An A/B intermediate will have C/C or T/T
3/ Firstly, as previously pointed out, Pekar et al's exclusion criterion of 'low read depth' is inconsistent with data from GISAID showing high read depth for the majority of the datasets
https://twitter.com/stevenemassey/status/1554910686626684931?s=20&t=6El9JQ9OgF6-iA_vhHAOcQ
4/ Many of the intermediate genomes are sequenced using ONT MinION/GridION/PromethION, a sequencing depth of > 60X is recommended for this platform
nature.com/articles/s4146…
Only 1 genome falls below this criterion (Table 1, in yellow)
nature.com/articles/s4146…
Only 1 genome falls below this criterion (Table 1, in yellow)


5/ Curiously, 'contamination' is used as an exclusion criterion. However, nowhere is any evidence presented of contamination. One way to do this is via haplogroup analysis of human mito genomes, to show more than one haplogroup present, which they fail to do

6/ 'Personal communications' are used to exclude 11 C/C and 3 T/T genomes. An 'L.Chen' is credited for the information that 11 intermediate C/C genomes from Sichuan and Wuhan are sequencing errors
7/ However, the identity of L.Chen is unclear. In addition, the C/C genomes (actually 12 not 11) from Sichuan and Wuhan were sequenced in different sequencing centers, so likely sequenced by two different people

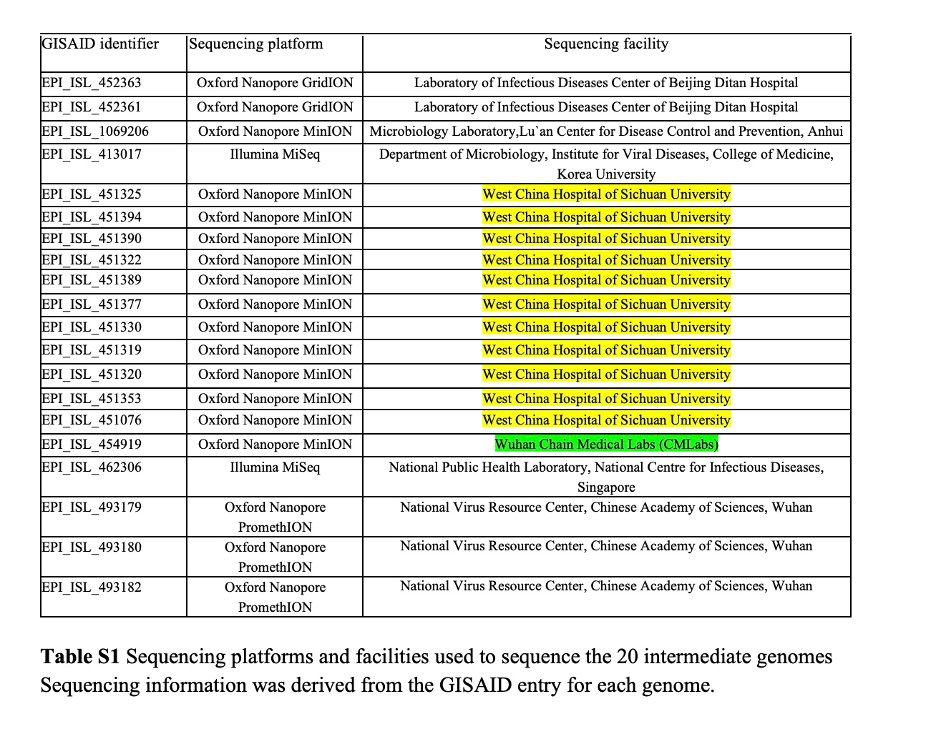
8/ Given L.Chen is explicitly linked by the authors to Sichuan 👇, then the Wuhan genome was sequenced by an unidentified person we term 'person X'. It is concerning that a C/C intermediate genome was excluded by a personal communication with an unidentified person

9/ One of the Sichuan C/C genomes (EPI_ISL_451320) excluded by Pekar et al. is actually used by NextStrain to root their phylogenetic tree (as an A/B root). This genome has a 1335X sequencing depth. Clearly @nextstrain do not concur that it is erroneous

10/ Pekar et al. exclude genomes from Singapore (EPI_ISL_462306) and South Korea (EPI_ISL_413017) that had raw data available, on the basis of low sequencing depth at positions 8782/28144 and 28144, respectively
However, we show that the Singapore genome is clearly a T/T genome
However, we show that the Singapore genome is clearly a T/T genome
11/ We do this by mapping the raw reads to the Hu-1 reference genome (which is C/T, lineage B)
Position 8782 has 12/12 T, while 28144 has 6/6 T
h/t @humblesci
This is clearly a T/T genome therefore
Position 8782 has 12/12 T, while 28144 has 6/6 T
h/t @humblesci
This is clearly a T/T genome therefore

12/ We also identify an additional intermediate T/T genome from Guangzhou (GZMU0025.capture, SRR13616010), that has the following SNVs when compared to Hu-1
The T8782 genotype is clear
h/t @humblesci Daoyu
The T8782 genotype is clear
h/t @humblesci Daoyu

13/ Remarkably, two C/C intermediate genomes from Beijing (2500X and 1850X sequencing depth) were excluded because 'no underlying data was available'.
This is hard to understand, and was selectively applied (it was not applied to the 787 remaining genomes used in their analysis)
This is hard to understand, and was selectively applied (it was not applied to the 787 remaining genomes used in their analysis)

14/ Puzzlingly, one T/T intermediate genome EPI_ISL_493182 was discarded even though it conformed to their (contentious) 10X read depth cutoff. Position 8782 is a consensus T nucleotide, with 19/29 reads T

15/ Pekar et al fail to explain how repeated sequencing errors can occur in the same positions 8782 and 28144 in multiple genomes
If sequencing depth were a problem in causing miscalls, there should be a significant number of unique SNVs in these genomes, which is not observed
If sequencing depth were a problem in causing miscalls, there should be a significant number of unique SNVs in these genomes, which is not observed
16/ 'Convergence' was used to exclude 7 intermediates that possessed A, B or A/B specific SNVs. However, 5 of these only possessed 1 A or B specific SNV. These could be true intermediates that picked up a A or B specific SNV by convergence
This caveat was not discussed
This caveat was not discussed

17/ To conclude, the exclusion of most of the 20 intermediate genomes from the analysis of Pekar et al. is untenable, and represents an unsurmountable problem for the conclusion of two zoonoses leading to the establishment of lineages A and B
18/ FYI @BillyBostickson @CharlesRixey @R_H_Ebright @JamieMetzl @franciscodeasis @lab_leak @jbloom_lab @pathogenetics @breakfast_dogs @BiophysicsFL @Biorealism
@threadreaderapp unroll
@threadreaderapp unroll
• • •
Missing some Tweet in this thread? You can try to
force a refresh










