I wanted to summarize current knowledge about origins of the #SARSCoV2 Omicron variant.
(This 🧵 doesn’t have anything new for people following topic closely, but I still get many questions about this, so am recapping current knowledge.)
(This 🧵 doesn’t have anything new for people following topic closely, but I still get many questions about this, so am recapping current knowledge.)
TLDR: there are now good reasons to favor explanation that Omicron largely evolved in chronic human infection(s), possibly w some compensatory evolution after re-entry into general human population.
No other proposed explanation look particularly convincing anymore.
No other proposed explanation look particularly convincing anymore.
To start, let’s review what was unusual about Omicron.
First, Omicron is on a very long branch from the rest of #SARSCoV2 phylogeny, indicating it has a lot of new mutations relative to anything before it (image below from nextstrain.org/nextclade/sars…).
First, Omicron is on a very long branch from the rest of #SARSCoV2 phylogeny, indicating it has a lot of new mutations relative to anything before it (image below from nextstrain.org/nextclade/sars…).

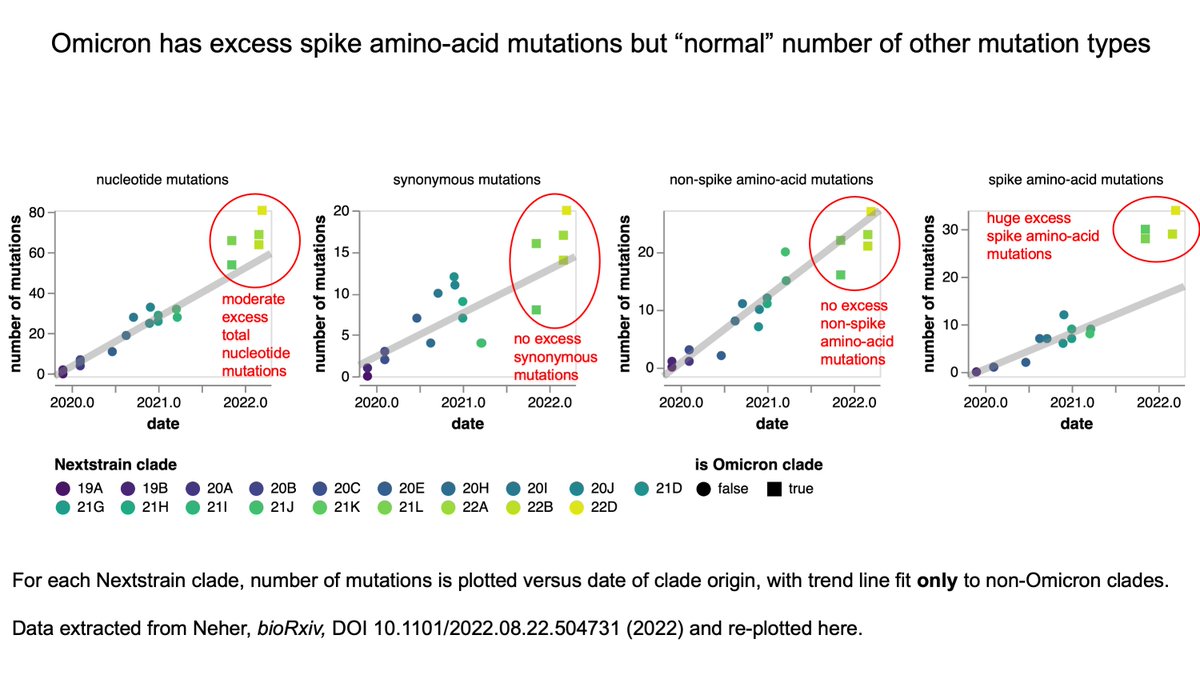
Second, for a virus of its “age”, Omicron has an excess of only one type of mutation: amino-acid mutations in spike.
Omicron has a “normal” number of synonymous mutations and non-spike amino-acid mutations for a virus that appeared when it did. See figure below:
Omicron has a “normal” number of synonymous mutations and non-spike amino-acid mutations for a virus that appeared when it did. See figure below:
Third, the excess amino-acid mutations in Omicron’s spike are concentrated in positions strongly targeted by human neutralizing antibodies. Many mutations in RBD, and within RBD they are at key antigenic positions (417, 446, 484, etc).
So Omicron emerged by process that fixed “normal” number of mutations for elapsed time in all of virus except spike, where there is strong enrichment of antibody-escape mutations.
Therefore, we can conclude there wasn’t elevated mutation rate, just elevated antibody selection.
Therefore, we can conclude there wasn’t elevated mutation rate, just elevated antibody selection.
To understand why this conclusion is justified, note that for *neutral* mutations, the rate of fixed mutations (substitutions) is just equal to the rate at which mutations appear: nature.com/scitable/knowl…
For a virus like #SARSCoV2, we expect many synonymous mutations to be roughly neutral. So if Omicron emerged by a process that elevated mutation rate, it would have excess synonymous mutations. But it doesn’t: it has normal number of synonymous mutations for virus of its age.
For mutations that aren’t neutral (like most amino-acid mutations), rate of fixed mutations depends on both mutation rate and selection on those mutations. If mutations are beneficial, then they will fix faster as viruses with those mutations will have a fitness advantage.
So Omicron evolved by process strongly favoring antibody-escape mutations in spike, but otherwise involving “typical” mutation rate & selection on non-spike proteins.
As most people reading this are probably already aware, we know of such process: chronic human infections.
As most people reading this are probably already aware, we know of such process: chronic human infections.
It is now extensively documented that chronic infections (typically immunocompromised patients) impose strong selection for mutations in spike, probably because virus is exposed to sub-neutralizing antibody for extended time w/o transmission bottlenecks:
https://twitter.com/jbloom_lab/status/1492974590460510210
For instance, in 2020 @DrJLi described an immunocompromised patient whose virus acquired 10 amino-acid mutations in spike over a ~140-day chronic infection:
https://twitter.com/jbloom_lab/status/1492974590460510210
A study by @GuptaR_Lab likewise described a chronic infection with multiple spike amino-acid mutations, and presciently suggested such infections could contribute to emergence of future #SARSCoV2 variants: nature.com/articles/s4158…
A study by @sigallab described extensive antibody escape via numerous amino-acid substitutions in spike in chronic infection of a HIV+ patient in South Africa:
sciencedirect.com/science/articl…
sciencedirect.com/science/articl…
A nice meta-analysis by @SternLab summarizes across many cases how chronic infections often lead to strong selection for antibody-escape mutations in spike that have characteristics of variants of concern:
nature.com/articles/s4159…
nature.com/articles/s4159…
Harm van Bakel & @VivianaSimonLab even caught the virus in the act, by identifying a highly mutated variant that appeared in a chronically infected patient and then transmitted to a few other people (although fortunately did not spread widely): medrxiv.org/content/10.110…
So for these reasons, evolution in chronically infected humans is certainly *consistent* with mutation properties of Omicron. (Although Omicron does have more spike mutations than observed in any single chronic infection yet studied.)
Another more subtle aspect of Omicron’s evolution is also consistent with strong antigenic during a chronic infection: when a protein is strongly selected for some specific trait (say antibody escape), it often impairs other important biochemical properties (like stability).
This impairment can happen because antibody-escape mutations themselves may come at a cost to other biochemical properties, & also because deleterious mutations can hitchhike w beneficial ones if they are close enough in primary sequence to stay in linkage disequilibrium.
And in fact, we see clear evidence that region of strongest antigenic selection during Omicron’s evolution (the RBD) acquired deleterious mutations that have been getting “repaired” during more recent evolution of Omicron subvariants.
One way they are getting repaired is simply by reversion. Several mutations in early Omicron variants have repeatedly reverted mutations (eg, at site 493 in Omicron’s RBD). A reversion is an obvious way to repair a defect caused by a hitchhiking deleterious mutation.
In addition, the RBD of earliest Omicron variants (eg, BA.1) was notably destabilized, and later Omicron variants have been acquiring secondary mutations that repair this biochemical defect. See below from @yunlong_cao:
https://twitter.com/yunlong_cao/status/1557449351165018112

Probably the strong selection for antibody escape in Omicron’s evolution in a chronic infection impaired RBD stability, which in turn hurt transmissibility--and so this defect has been getting repaired during subsequent compensatory evolution in the general population.
Now I want to address other theories initially proposed for Omicron’s origins: (1) evolution in isolated human population, (2) jump from animal reservoir, (3) lab accident, (4) mutagenic agent like molnupiravir.
None of these explain the data well.
None of these explain the data well.
Evolution in isolated human population: (a) we don’t know of any populations sufficiently large to sustain extended transmission while remaining totally isolated; (b) evolution in isolated population can’t explain elevated rate of antibody-escape mutations in spike.
Jump from animal reservoir: there is no evidence that #SARSCoV2 picks up a dramatic excess of antibody-escape mutations in spike when it evolves in animals.
In fact, antigenic evolution is likely to be *slower* in animals than humans: researchsquare.com/article/rs-136…
In fact, antigenic evolution is likely to be *slower* in animals than humans: researchsquare.com/article/rs-136…
Lab accident: while circumstantial evidence makes this a plausible theory for origins of #SARSCoV2 itself in Wuhan, this evidence doesn’t extrapolate to Omicron. South Africa (where Omicron identified) has plenty of HIV+ chronic infections.
Also, engineering of spike for antibody escape would not explain how Omicron also ended up with “normal” number of new mutations elsewhere in genome.
Additionally, this paper (zenodo.org/record/6904363…) arguing for lab origin of Omicron applies statistics that “neglect the effect of natural selection,” which is a poor assumption because Omicron’s spike clearly evolved under strong natural selection.
Finally, mutagenic drug such as molnupiravir would lead to excess mutations throughout genome, not just amino-acid mutations in spike.
Extended Fig 1 of this paper (biorxiv.org/content/10.110…) shows example #SARSCoV2 hypermutation: lots of mutations everywhere. That's not Omicron.
Extended Fig 1 of this paper (biorxiv.org/content/10.110…) shows example #SARSCoV2 hypermutation: lots of mutations everywhere. That's not Omicron.
Overall, I hope this thread recaps why chronic human infections are clearly best explanation for emergence of Omicron.
This doesn’t mean we shouldn’t keep the other possible explanations in mind as concerns for future, but they are unlikely to have contributed to Omicron.
This doesn’t mean we shouldn’t keep the other possible explanations in mind as concerns for future, but they are unlikely to have contributed to Omicron.
• • •
Missing some Tweet in this thread? You can try to
force a refresh