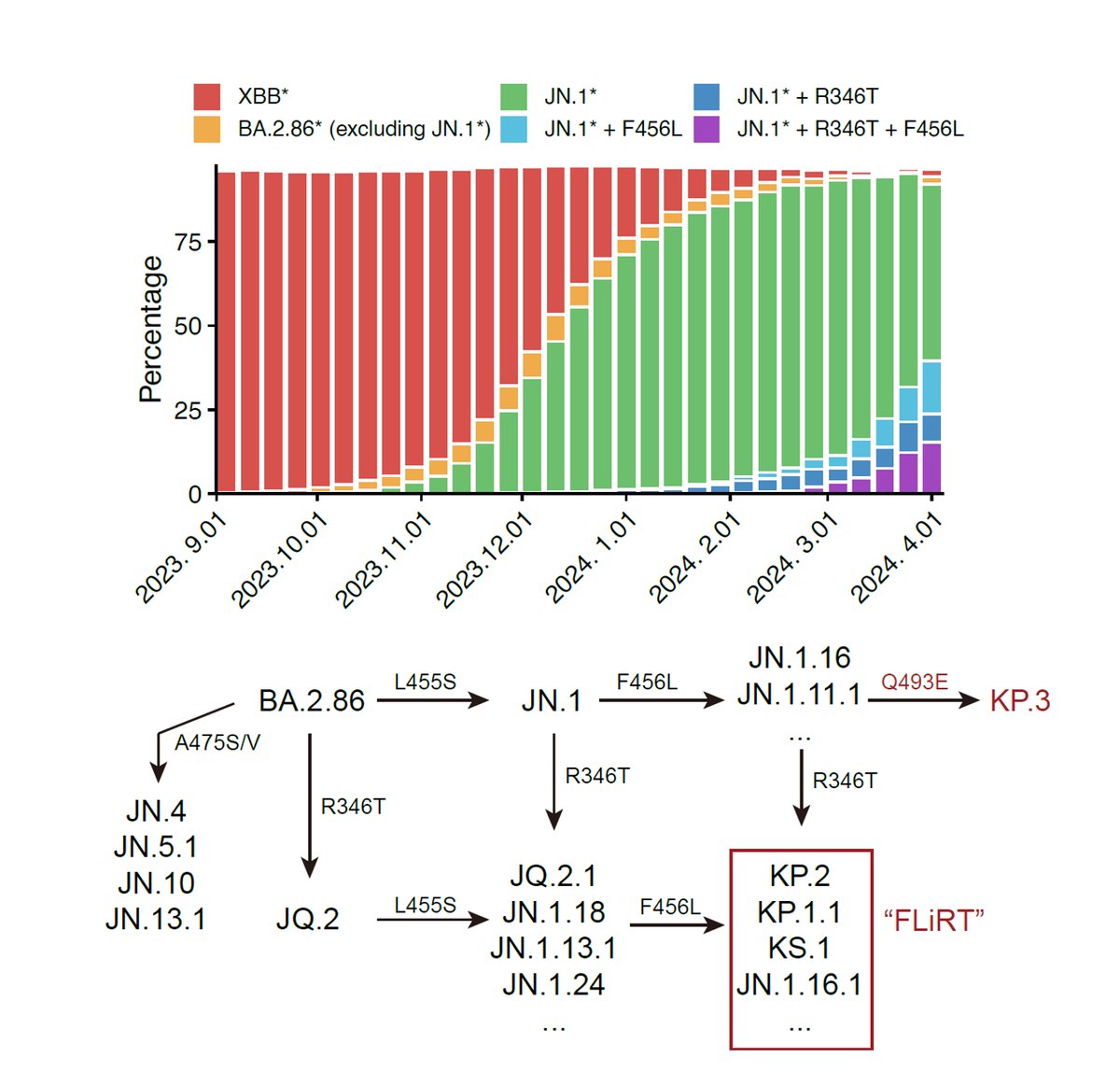
Sharing our investigation on the unprecedented convergent RBD evolution of BA.2.75 and BA.5 on sites including 346, 356, 444-446, 450, 460, 486, which have generated highly concerning variants such as BA.2.75.2, BR.1, BJ.1, and BQ.1.1. (1/n)
biorxiv.org/content/10.110…
biorxiv.org/content/10.110…
In this paper, we tried to solve the following three questions:
1) How immune evasive could these variants be?
2) Why do they evolve mutations on these converging sites?
3) What could this convergence evolution finally lead to? (2/n)
biorxiv.org/content/10.110…
1) How immune evasive could these variants be?
2) Why do they evolve mutations on these converging sites?
3) What could this convergence evolution finally lead to? (2/n)
biorxiv.org/content/10.110…
As many have noticed, recent evolution of Omicron has led to numerous subvariants that exhibit high growth advantages over BA.5. Interestingly, mutations on their receptor-binding domain (RBD) converge on several hotspots, including R346, R356, K444, L452, N460 and F486. (3/n) 

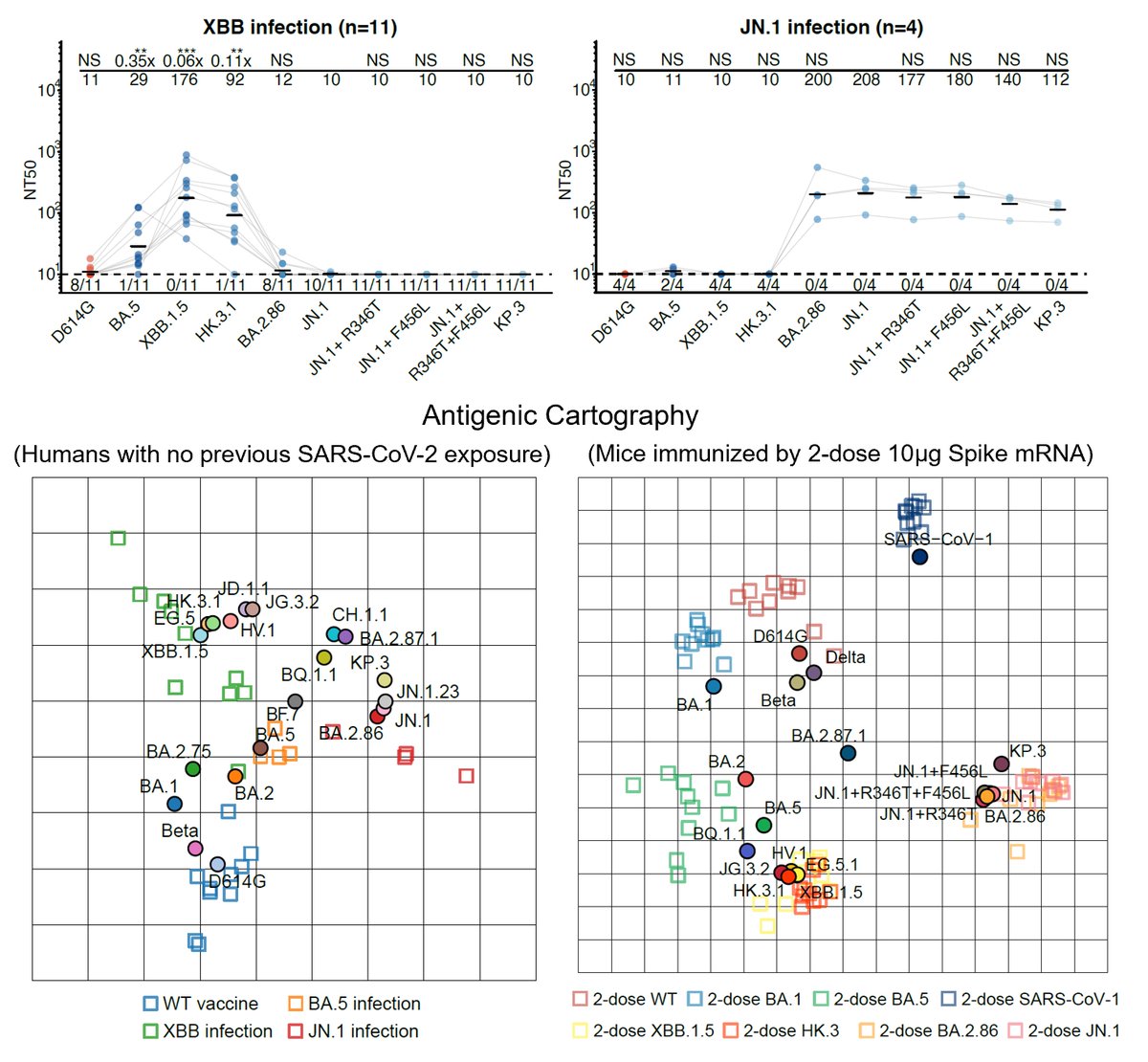
First, we tested the antibody evasion capability of these variants. Therapeutic antibodies are not very effective against those convergent variants. Bebtelovimab doesn't work against variants carrying K444N/M/T or V445P. Evusheld is also heavily escaped. (4/n)

As for plasma, BA.2.75.2 exhibits the most significant reduction in NT50, even for BA.5 breakthrough-infected convalescents. Almost 10-fold difference compared to BA.5 against plasma from BA.5 convalescents. Will update results on 6~8 new variants, such as BQ.1.1 next week. (5/n)


Proved by pseudovirus neutralization assays using soluble hACE2, we found that all of the tested variants exhibited sufficient hACE2 affinity, higher than that of D614G, indicating they all have a chance to circulate. (6/n)

Next, we investigated why Omicron suddenly started to evolve convergently. In short, we believe this is linked to humoral immune imprinting. Similar to BA.1 breakthrough infection, we showed BA.2/BA.5 breakthrough infection also mainly recalls previous WT-induced memory. (7/n)

We isolated RBD-antibodies from BA.2/5 convalescent plasma, measured their neutralization, determined their epitope and escaping mutations using high-throughput DMS. Integrated with our previous data, we finally got a DMS dataset containing 3051 RBD-reactive antibodies. (8/n)

Importantly, BA.5 breakthrough infection exhibited further enrichment of non-neutralizing epitopes (E2.2/E3/F1, 63%) and less diversified NAb epitopes (less B/D1/E2.2 and more D2). The reduced B/D1/E2.2 antibodies is due to the F486V and L452R mutations. (9/n)
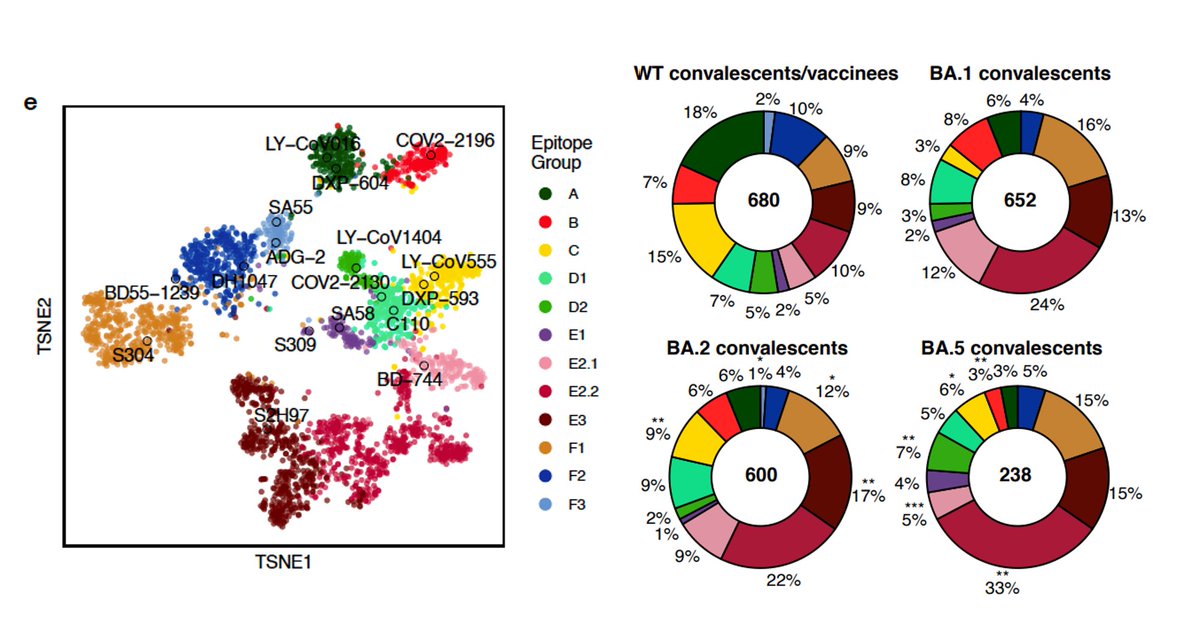
These observation means that due to immune imprinting, BA.5 breakthrough infection mainly recalls previous memory and rarely produces mAbs with new epitopes. This causes a significant reduction of NAb epitope diversity and an increased proportion of non-neutralizing mAbs. (10/n)
To better visualize the above observation, we modified the algorithm by @jbloom_lab and showed the immune presure elicited by BA.2/BA.5 breakthough infection. This strikingly shows the reduced NAb diversity and concentrated immune pressure for BA.5 infection than BA.2. (11/n)

By integrating data of neutralization, ACE2-binding, RBD-stability, and codon-usage (method detailed described in the manuscript), we can accurately identify sites conferring high immune pressure and infer mutations preferred to appear. (12/n)
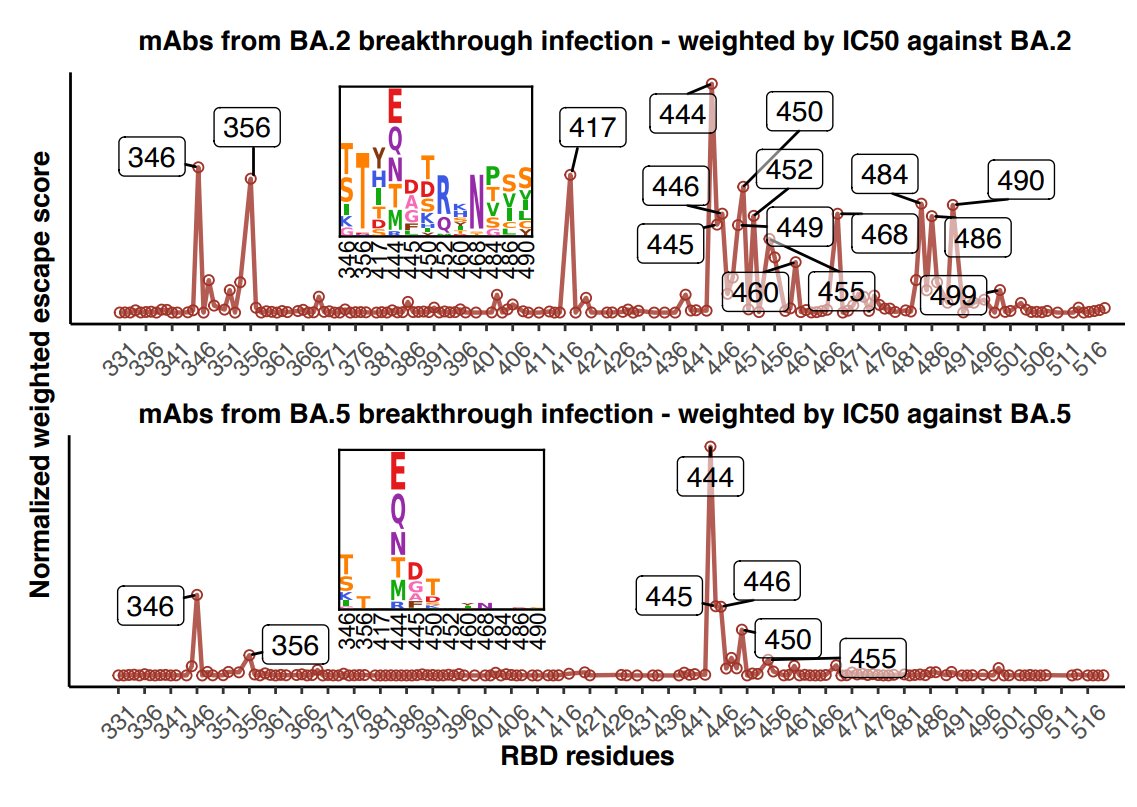
Check how well this model aligns with the convergent mutations! R346T/S/I, K356T, K444N/T/M, N450D, L452R, N460K, F486S/V/I, and F490S. (13/n)

The above analyses suggest: due to immune imprinting, BA.5 breakthrough infection caused significant reductions of nAb epitope diversity and increased proportion of non-neutralizing mAbs, which in turn concentrated immune pressure and promoted the convergent RBD evolution. (14/n)
Lastely, we want to know if this convergence evolution keeps on going, what would it finally lead to? We started by calculating the convergent RBD evolution trend of BA.2.75 and BA.5. The result fits very well with the emerging variants, such as BA.2.75.2, BR.1, BQ.1.1. (15/n)

We checked whether these indicated mutations could work together. We chose ~200 BA.2-effective NAbs from distinct epitope groups and tested their neutralization against those mutations. Results suggest the mutations synergize very well and could escape almost all RBD-NAbs. (16/n)

Then we constructed pseudoviruses carrying stacked convergent mutations to find the destination of the convergence, and tested the neutralization of plasma and NAb drugs. NAb drugs are escaped as expected, except SA55. All of the mutants exhibit sufficient ACE2-binding. (17/n)

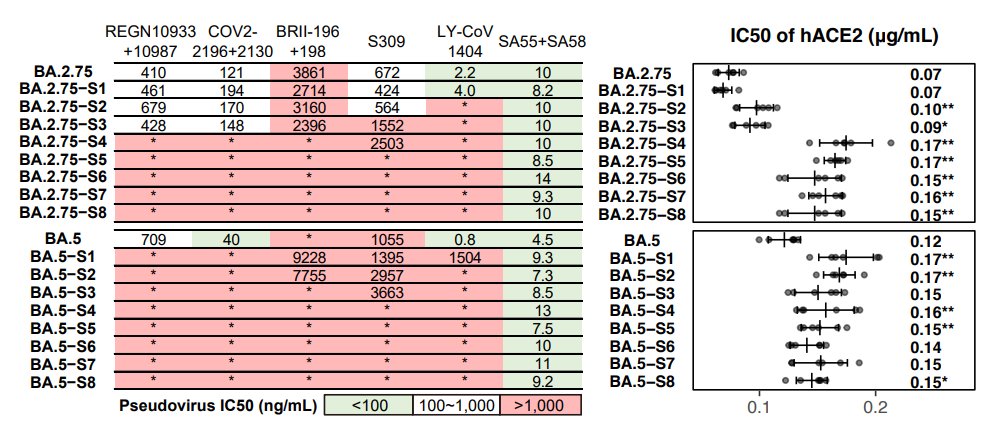
In BA.2.75 derivatives, F486V confers the most striking drop in NT50 for WT/BA.1/BA.2 plasma but R346T for BA.5 plasma. We also found BA.2.75-S4 is enough to eliminate the neutralization of most plasma samples, and NTD mutations are necessary to escape BA.2/5 plasma. (18/n)
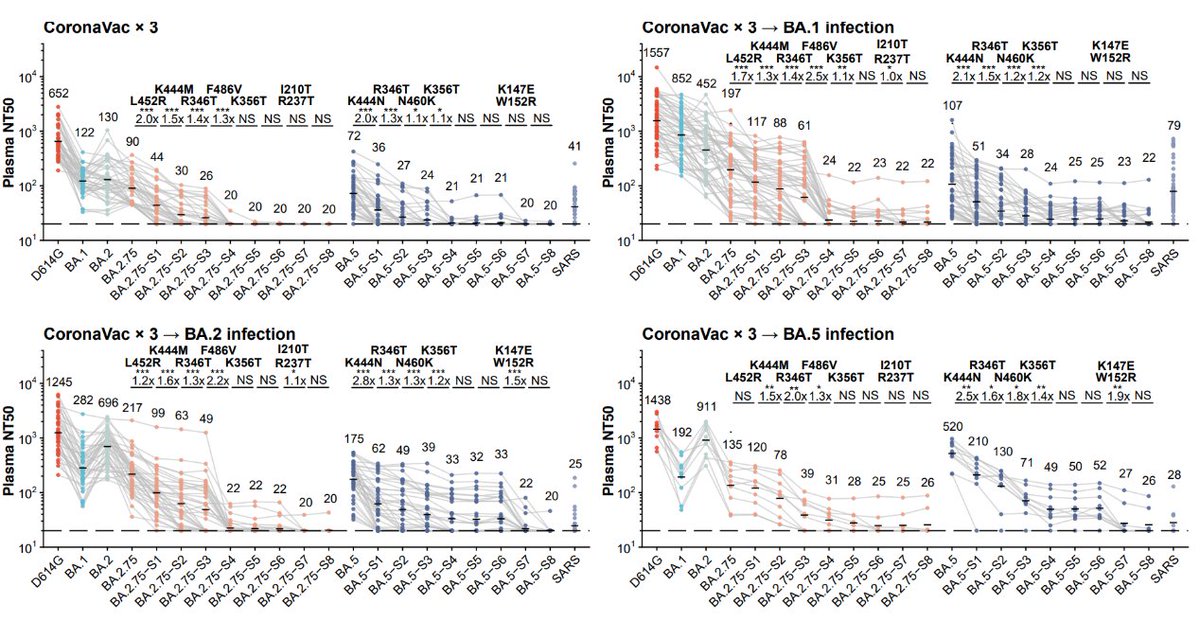
Note how close BA.5-S4 (BA.5+R346T+N417T+K444N+N460K) is compared to the recent BQ.1.1. They will have similiar evasion capability and the results already show how evasive BQ.1.1 would be. This also proves our prediction is on track. (19/n)


Together, our results suggest natural infection or vaccine boosters using BA.5 may not provide sufficiently broad protection. Broad-spectrum vaccines and NAb drugs should be developed and our constructed convergent mutants could help examine their effectiveness in advance. (20/n)
We will update information on BA.2.3.20,BN.1,BA.4.6.1,BQ.1,BQ.1.1,BA.2.10.4, BN.2.1 next week. This work is greatly inspired by @jbloom_lab, and we will share all the DMS and neutralization data so everyone could play with it using Jessi’s @jbloom_lab calculator. (21/n)
• • •
Missing some Tweet in this thread? You can try to
force a refresh