
With 3 more days of data, here's a look at how far various collections of immune escape variants have progressed so far.
I now count S:493 as a key mutation, so what used to be >=5 (Pentagon) is now >=6.
The more RBD mutations, the faster the growth.
cov-spectrum.org/explore/World/… 1/
I now count S:493 as a key mutation, so what used to be >=5 (Pentagon) is now >=6.
The more RBD mutations, the faster the growth.
cov-spectrum.org/explore/World/… 1/
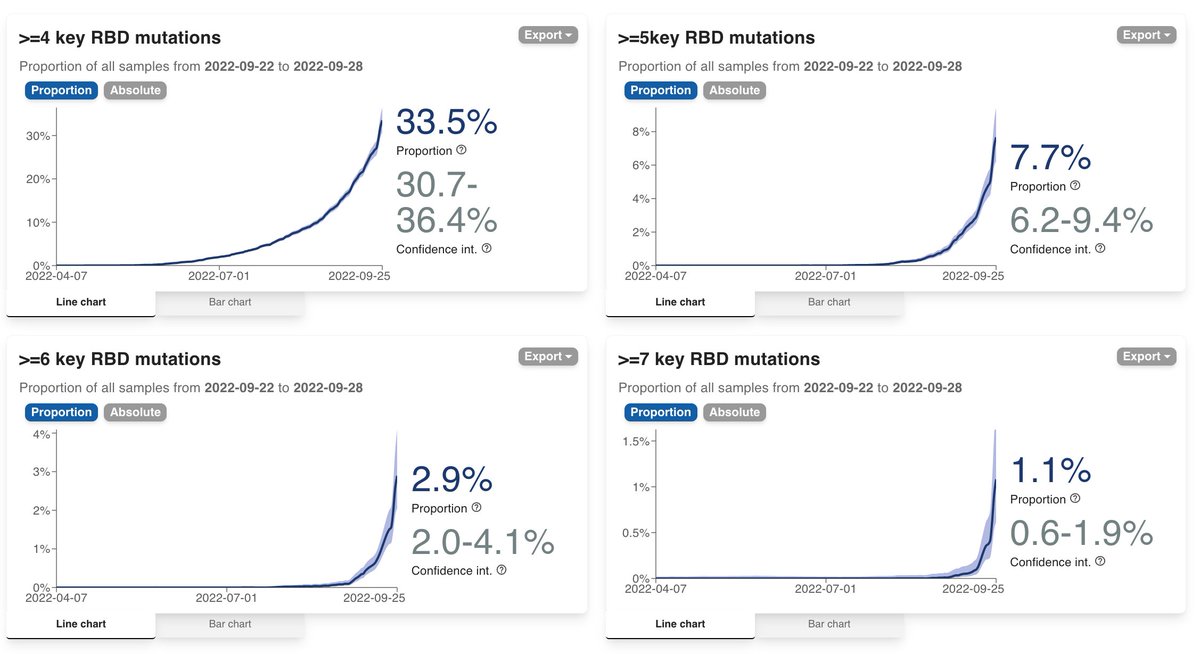
Looking at the data like this, we reduce the complexity behind numerous emerging variants into 4 buckets of increasing escape. This is not perfect as what's in a bucket is not totally the same, but it solves the problem of having to remember BQ.1.1, BA.2.75.2, XBB etc. 2/

If you want to connect this way of looking at things with various lineage names you may have heard of, this is a summary
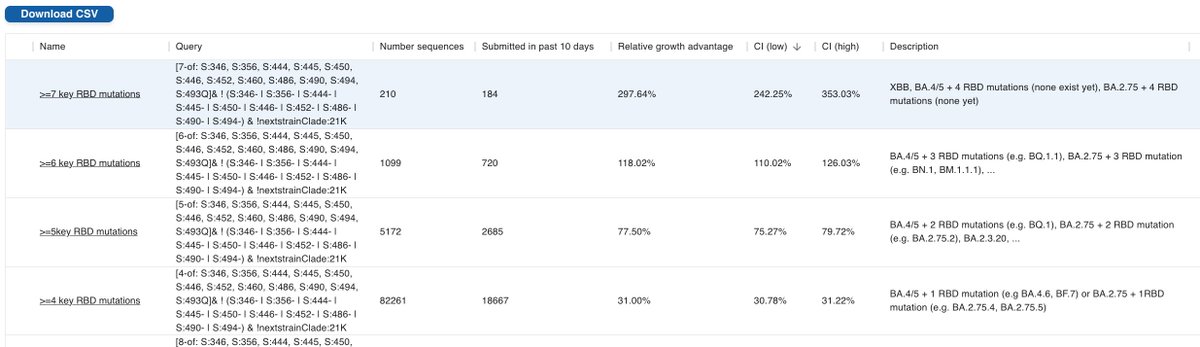
Level 3 (not shown as "boring") = BA.4/5, BA.2.75 = BA.2 + 3 RBD muts
Level 4 = BA.4/5 + 1 RBD mut (e.g. BA.4.6, BF.7) or BA.2.75 + 1 RBD mut (BA.2.75.5) 3/
Level 3 (not shown as "boring") = BA.4/5, BA.2.75 = BA.2 + 3 RBD muts
Level 4 = BA.4/5 + 1 RBD mut (e.g. BA.4.6, BF.7) or BA.2.75 + 1 RBD mut (BA.2.75.5) 3/

Level 5 = BA.4/5 + 2 (BQ.1, ...) , BA.2.75 + 2 (BA.2.75.2, BY.1, ...)
Level 6 = BA.4/5 + 3 RBD mut (e.g. BQ.1.1) or BA.2.75 + 3 RBD mut (BN.1, BM.1.1.1, ...)
Level 7 = XBB or BA.4/5 + 4 RBD mut (none so far) or BA.2.75 + 4 RBD mut (none so far) 4/
Level 6 = BA.4/5 + 3 RBD mut (e.g. BQ.1.1) or BA.2.75 + 3 RBD mut (BN.1, BM.1.1.1, ...)
Level 7 = XBB or BA.4/5 + 4 RBD mut (none so far) or BA.2.75 + 4 RBD mut (none so far) 4/
Each key RBD mutation that's added increases the growth advantage. But it also takes longer for that combination to evolve, so higher levels start out at lower share - to catch up later
While Level 3 (vanilla BA.4/5) still dominate in North America/Europe, that's not for long 5/
While Level 3 (vanilla BA.4/5) still dominate in North America/Europe, that's not for long 5/
S:346T, which is in most of level 4, is on track to become dominant by the end of October as I predicted 7 weeks ago here: github.com/neherlab/SARS-…
(note any higher level variant is automatically a member of lower levels, hence level 5 (BA.2.75.2) is also level 4) 5/
(note any higher level variant is automatically a member of lower levels, hence level 5 (BA.2.75.2) is also level 4) 5/


But higher levels are catching up very fast. Given the rapid growth in particular of level 6 variants (BQ.1.1, BN.1, BM.1.1.1, XBB) a variant driven infection wave is inevitable by the end of November, possibly as early as early November. 6/
As a case in point for using level buckets rather than individual lineages when evaluating the variant situation: the level 6 variants mentioned above (BQ.1.1, BN.1, BM.1.1.1, XBB) each only make up between 10-35%. If you look at only one of them, you'll miss most! 7/

Kudos to @ChaoranChen_ for implementing collections in @GenSpectrum, allowing these complex queries to be listed. The idea for collections came from a call that also included @shay_fleishon's team, and @TanjaStadler_CH.
Link to the "bucket" collection: cov-spectrum.org/collections/54 8/
Link to the "bucket" collection: cov-spectrum.org/collections/54 8/

If you prefer to have each lineage broken out individually, collection 24 maintained by @siamosolocani is the one to go for.
Note that it's important to not take the growth advantages at face value. Use the low confidence interval instead: cov-spectrum.org/collections/24 9/
Note that it's important to not take the growth advantages at face value. Use the low confidence interval instead: cov-spectrum.org/collections/24 9/
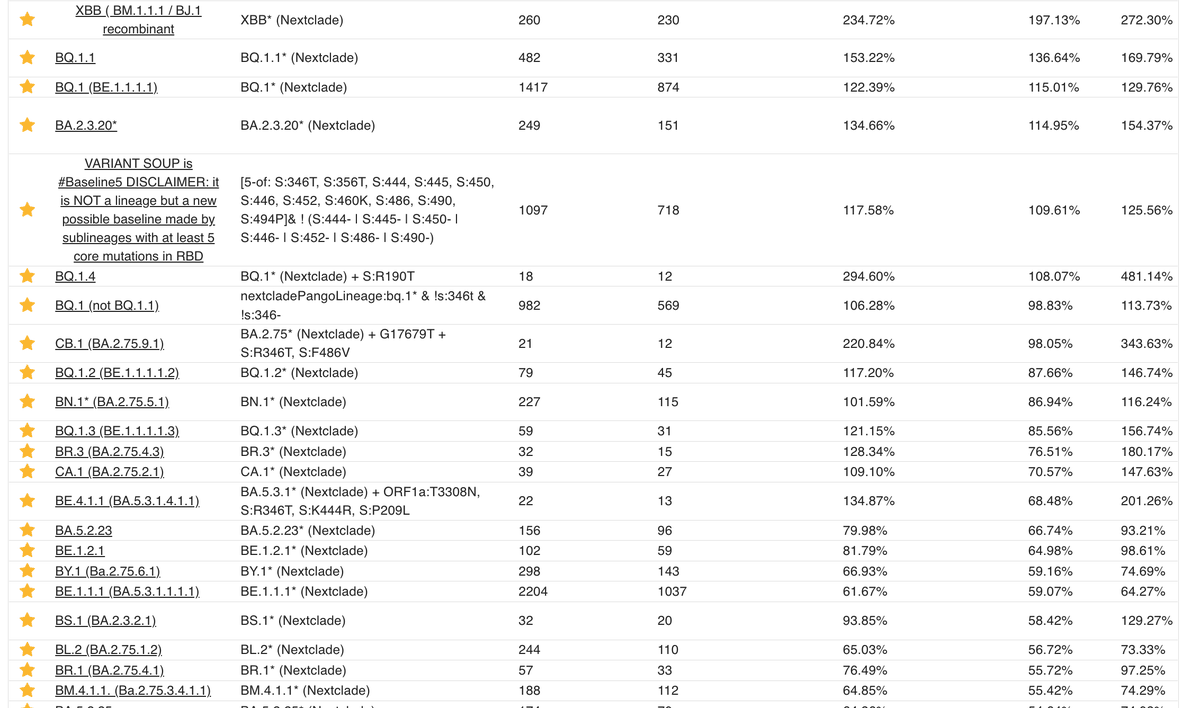
Beware that the proposed simplification into RBD mutation count levels ignores a lot of details that are important for individual variant success.
It only looks at mutations at 12 sites, when in fact very many positions matter, like the N-terminal domain of Spike. 10/
It only looks at mutations at 12 sites, when in fact very many positions matter, like the N-terminal domain of Spike. 10/
But for now, this seems to be a useful additional way of looking at the data and simplifying it to not be so confusing and still useful conceptually.
For the gold standard of growth advantage estimates, please have a look at @TWenseleers's analyses. 11/
For the gold standard of growth advantage estimates, please have a look at @TWenseleers's analyses. 11/
@GenSpectrum is quick and dirty, and definitely useful, but it's important to not take these values at face value when there is not enough data yet.
In contrast, Tom's estimates are much more robust and less biased.
In contrast, Tom's estimates are much more robust and less biased.
https://twitter.com/TWenseleers/status/1577810021592834049?s=20&t=J8Be5-VuNf_KbtW3q22V2g12/
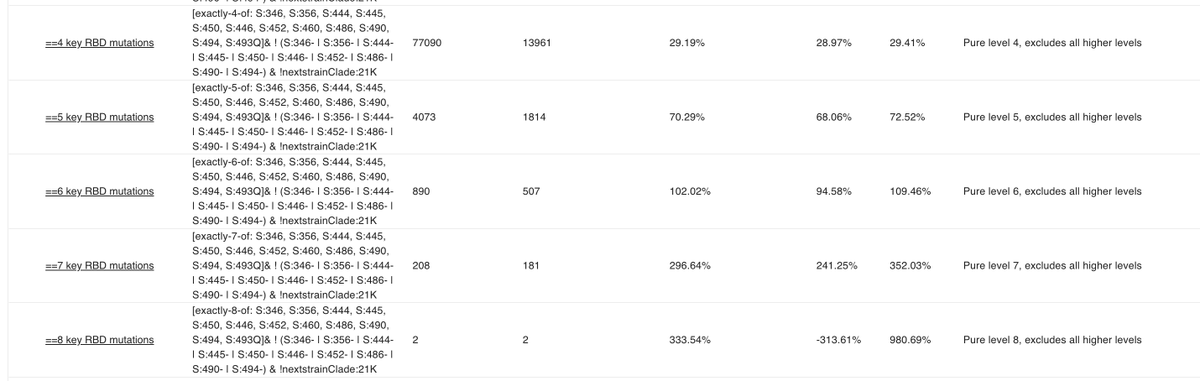
Prompted by a tweet from @BarakRaveh I added "pure" level X queries to collection 54:
A pure level 4 query shows everything with exactly 4 RBD mutations on top of BA.2 instead of 4 or more.
This is suitable for addition: level 4 + level 5 etc
A pure level 4 query shows everything with exactly 4 RBD mutations on top of BA.2 instead of 4 or more.
This is suitable for addition: level 4 + level 5 etc
https://twitter.com/BarakRaveh/status/1578305905153540096?s=20&t=oHQPHYe4QTEFWyuB3a_I-w
• • •
Missing some Tweet in this thread? You can try to
force a refresh







