You have to hand it to Lex Fridman. His grift is not an amateur job. Take his Twitter photo. A professor standing in front of a blackboard with some math. Right? 

This photo (see RHS of image below) is from what he calls his "MIT course" on Deep Learning for Self-Driving Cars. Sounds like good stuff. CS, math, self driving cars. #broheaven. So what is the problem? He is standing in front of the blackboard.

Well first of all, this was an MIT IAP class. IAP is a short period in January when students get to take fun classes on various topic that can be taught by anyone (many by students). I once sat in on a brain dissection. You can learn how to count cards. web.mit.edu/willma/www/mit…
The blackboard Lex is standing in front of has basic calculus left over from an actual real MIT calculus class. It has nothing to do with what he is "teaching". The stuff he is presenting is a joke. You can listen in to some of his rubbish here:
He clearly doesn't know what he's talking about. This explanation of L1 and L2 is 😭 He is standing in front of the formula 1-cos(2θ). I doubt he could tell you what the cosine of an angle is.
I watched all this crap so you don't have to.
I watched all this crap so you don't have to.
I'm not the first to figure this out... news.ycombinator.com/item?id=272553…
Everybody knows.
Everybody knows.
Lex doesn't quite lie, but obviously he is far from telling the whole truth. His "research position" is a whole other bunch of bull (for another time). And he uses this MIT mirage to great advantage, creating the perception that he is effectively a professor there.

By now people just take it for granted that he is an MIT prof. And he nurtures that myth. As I said, gotta hand it to him.

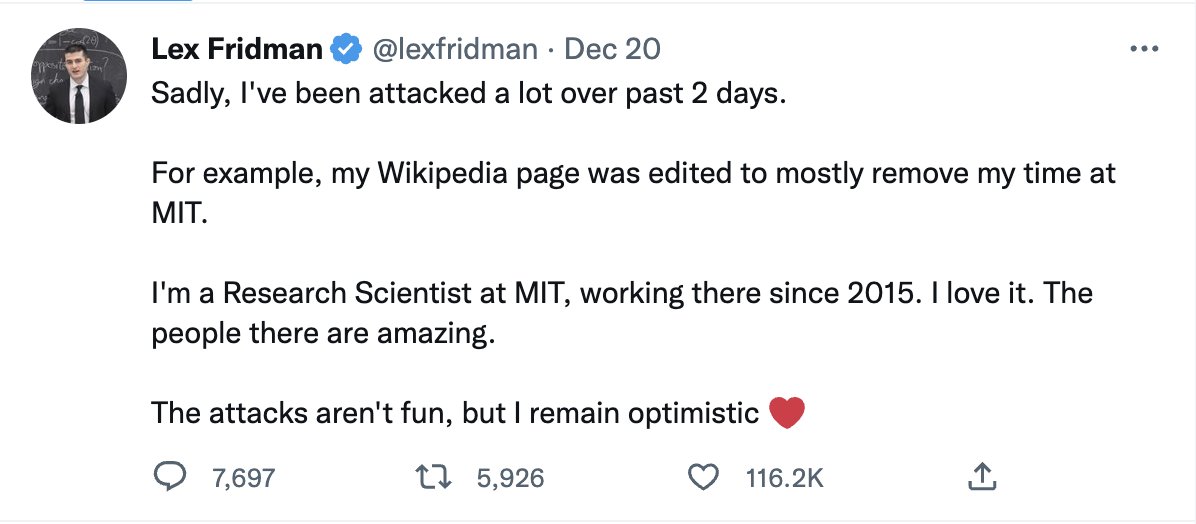
Full disclosure: Lex blocked me after I questioned why (with very few exceptions) he interviews only men.
https://twitter.com/lpachter/status/1303469780511285248?s=20&t=QcRl7USu8f8FsAJSjJuCaA
• • •
Missing some Tweet in this thread? You can try to
force a refresh