New preprint! 🥳
biorxiv.org/content/10.110…
Led by @aledjohnparry & @ChristelKrueger we asked why #DNAmethylation and oxidation often target overlapping genomic regions at the exit of #pluripotency.
Great collaboration with @Tim_Lohoff, @StevenWingett & @stefanschoenfe1.
🧵👇
biorxiv.org/content/10.110…
Led by @aledjohnparry & @ChristelKrueger we asked why #DNAmethylation and oxidation often target overlapping genomic regions at the exit of #pluripotency.
Great collaboration with @Tim_Lohoff, @StevenWingett & @stefanschoenfe1.
🧵👇
Excellent work by @SchubelerLab, @rulandsgroup, Jaenisch lab, Meissner lab, @CarellThomas and others has shown that DNA methylation turns over genome wide as cells exit naïve pluripotency, especially at enhancer elements.
We discussed this here: rdcu.be/b8bfM
We discussed this here: rdcu.be/b8bfM

But what is the function of methylation turnover?
To find out, we modelled pluripotency exit in vitro (transitioning ESCs to EpiLCs) and performed a thorough molecular characterisation in wild type cells and in cells lacking DNA methylation (Dnmt TKOs) or oxidation (Tet TKOs).
To find out, we modelled pluripotency exit in vitro (transitioning ESCs to EpiLCs) and performed a thorough molecular characterisation in wild type cells and in cells lacking DNA methylation (Dnmt TKOs) or oxidation (Tet TKOs).
This was a lot of data to wrangle (>300 datasets! 🥵), so we developed new computational approach.
We clustered promoter - promoter interacting region (PIR) pairs (identified by PCHi-C) using the expression, interaction strength and epigenetic information from both regions.
We clustered promoter - promoter interacting region (PIR) pairs (identified by PCHi-C) using the expression, interaction strength and epigenetic information from both regions.

This identified pairs of elements including promoters interacting with active enhancers, primed enhancers or poised enhancers.
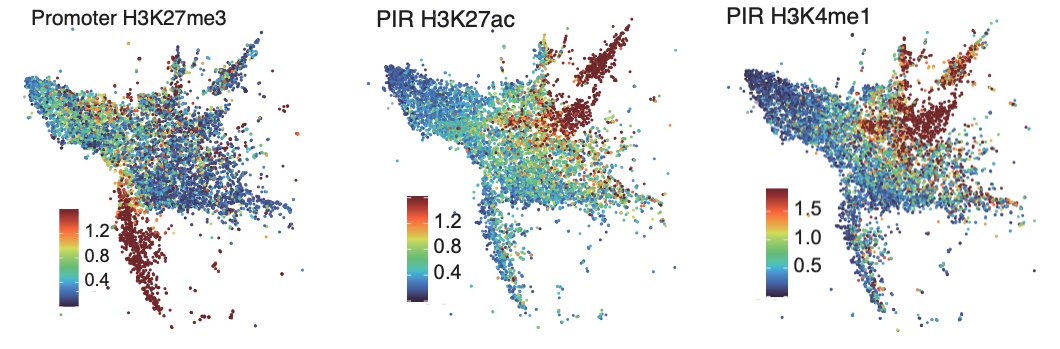
So what happens at enhancers in Dnmt and Tet TKO epiblast-like cells?
Excitingly we found that H3K4me1 is depleted from enhancers in both knockouts, but H3K27ac is reduced only in the Tet TKOs.
Excitingly we found that H3K4me1 is depleted from enhancers in both knockouts, but H3K27ac is reduced only in the Tet TKOs.

So we reasoned that methylation turnover is required for H3K4me1 deposition at active enhancers.
We have previously identified WRD5, a member of the MLL3/4 containing WRAD complex, as a strong binder of 5fC - this is one possible mechanism.
doi.org/10.1186/gb-201…
We have previously identified WRD5, a member of the MLL3/4 containing WRAD complex, as a strong binder of 5fC - this is one possible mechanism.
doi.org/10.1186/gb-201…

Is methylation turnover important for enhancer priming too?
To find out, we differentiated TKO ESCs into embryoid bodies (EBs). In Tet TKO EBs, activation of genes associated with the blood lineage was especially perturbed.
To find out, we differentiated TKO ESCs into embryoid bodies (EBs). In Tet TKO EBs, activation of genes associated with the blood lineage was especially perturbed.
Excitingly we could already see defects at primed enhancers associated with key blood transcription factor (TF) genes in Tet and Dnmt TKO epiblast like cells, long before these genes are expressed.
Primed enhancers lost H3Kme1 . . .
Primed enhancers lost H3Kme1 . . .

. . . and interaction strength between these primed enhancers and their associated genes was often reduced.
Thus TET and DNMT activity (and therefore methylation turnover?) are both important for priming of enhancers associated with genes encoding blood specific TFs.
Thus TET and DNMT activity (and therefore methylation turnover?) are both important for priming of enhancers associated with genes encoding blood specific TFs.

For example, two blood TF genes, Klf1 and Lyl1, share a number of primed enhancers.
These enhancers and the interactions between them were clearly perturbed in Tet TKO epiblast-like cells, potentially explaining why the genes aren’t upregulated upon differentiation.
These enhancers and the interactions between them were clearly perturbed in Tet TKO epiblast-like cells, potentially explaining why the genes aren’t upregulated upon differentiation.

Perhaps DNA methylation turnover at enhancers can explain the paradoxical finding that mutations in DNMT and TET genes are both causally linked to blood cancer. Enhancer activity is disrupted if either methylation or oxidation is perturbed.
We’d love to know your thoughts!
We’d love to know your thoughts!
• • •
Missing some Tweet in this thread? You can try to
force a refresh