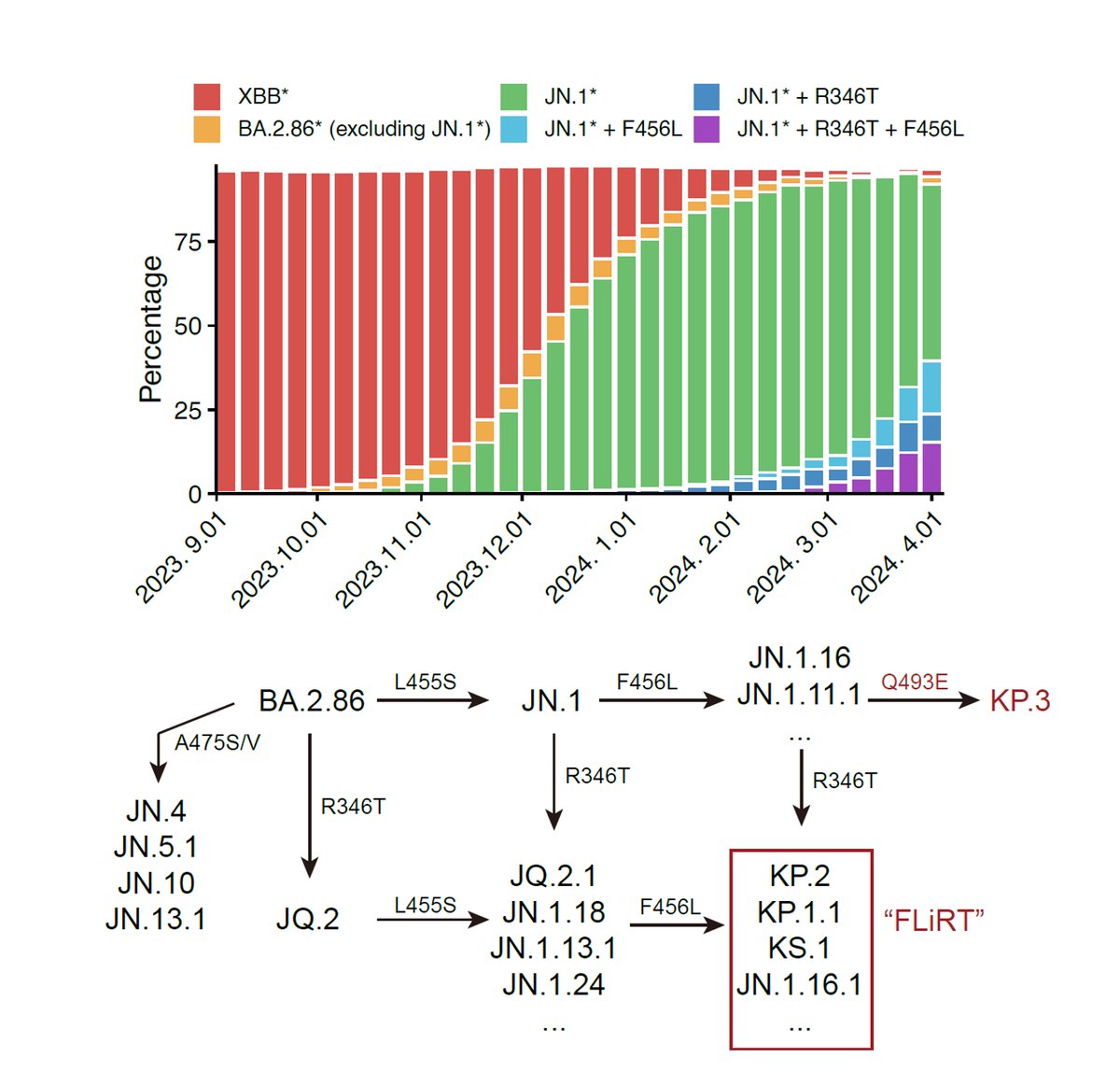
Recently, many fast-growing XBB lineages have gained RBD mutations on K478, such as VOI XBB.1.16 (K478R), XBB.2.3.5 (K478N), XBB.2.3.4 (K478Q). Also, many XBB* have independently obtained F456L, like FD.1.1, FE.1, XBB.1.5.10. In this thread I'll briefly discuss these mutations. 

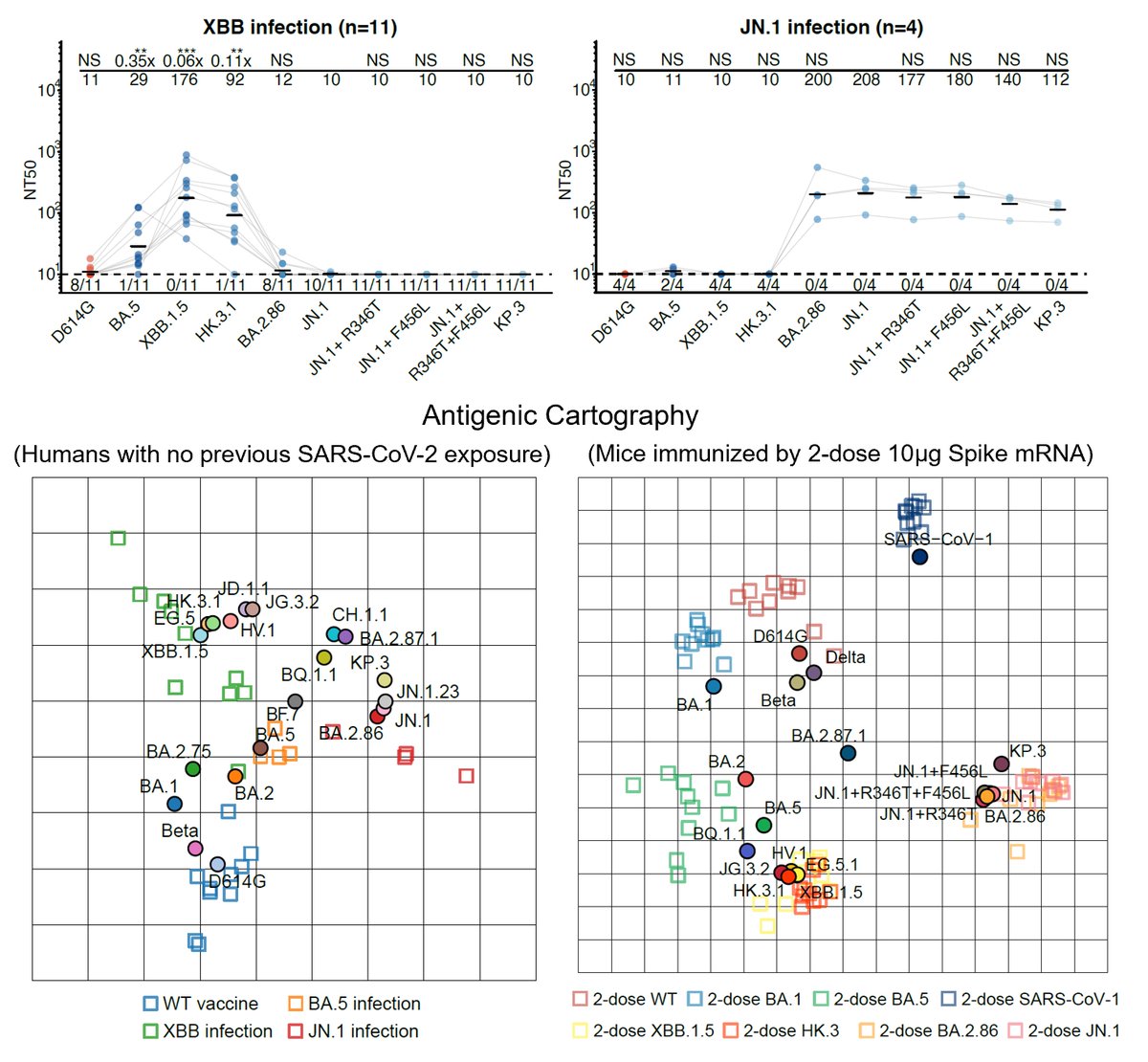
Like the results by Kei @SystemsVirology, we found XBB.1.16 and XBB.1.5 have comparable immune evasion capabilities in the serum tested. The ACE2 binding affinity of XBB.1.16 and XBB.1.5 is also similar. In contrast, F456L brings additional immune evasion but lowers ACE2 binding.

F456L escapes XBB.1.5-effective class I mAbs. These mAbs are quite abundant in various immune backgrounds, such as people who experienced BA.5 breakthrough infections or repeated Omicron infections. Those that are developing RBD-targeting mAb drugs should pay attention to F456L

K478 mutations can also escape mAbs whose epitope centered around 478, as revealed by DMS. However, these mAbs are pretty rare in the cohorts we examined, thus we don't see additional serum immune evasion by XBB.1.16. Note that these mAbs are Omicron-specific (cannot bind WT RBD)
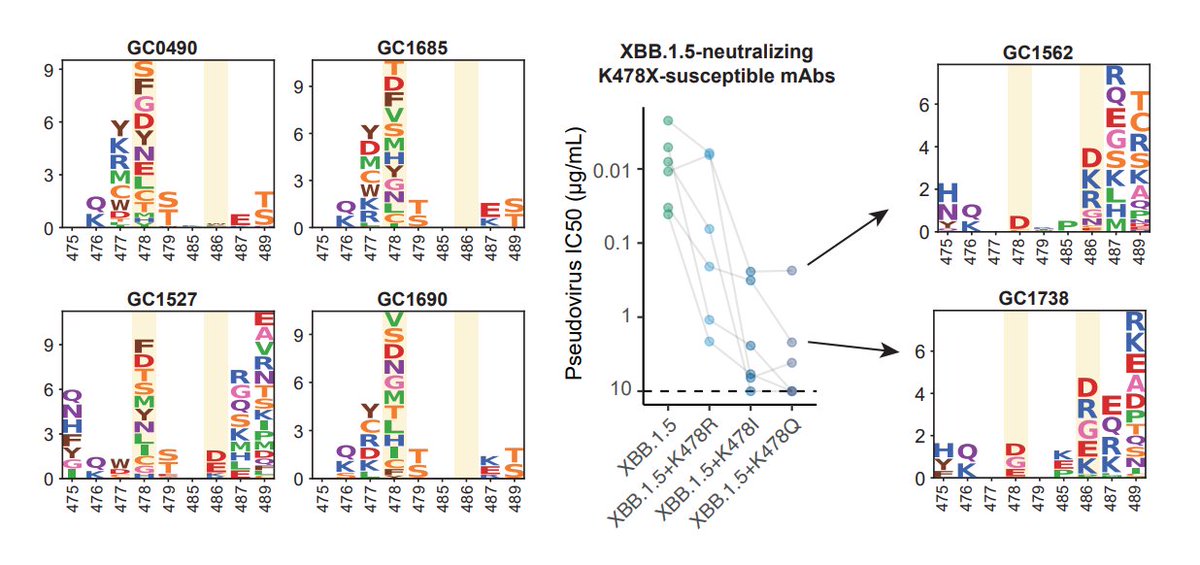
The lack of strengthened immune evasion or ACE2 binding cannot explain why K478 mutations are constantly popping out in fast-growing lineages. This contradiction might be due to the fact that we haven't captured the real immune background that introduced those 478 mutations.
One potential background that can give rise to K478X is repeated BA.5/BQ.1.1/XBB exposures. This is because F486 would mask the immunogenicity of K478 (so F486 must mutate), and two Omicron stimulation is needed to alleviate immune imprinting for Omicron-specific mAbs to appear.
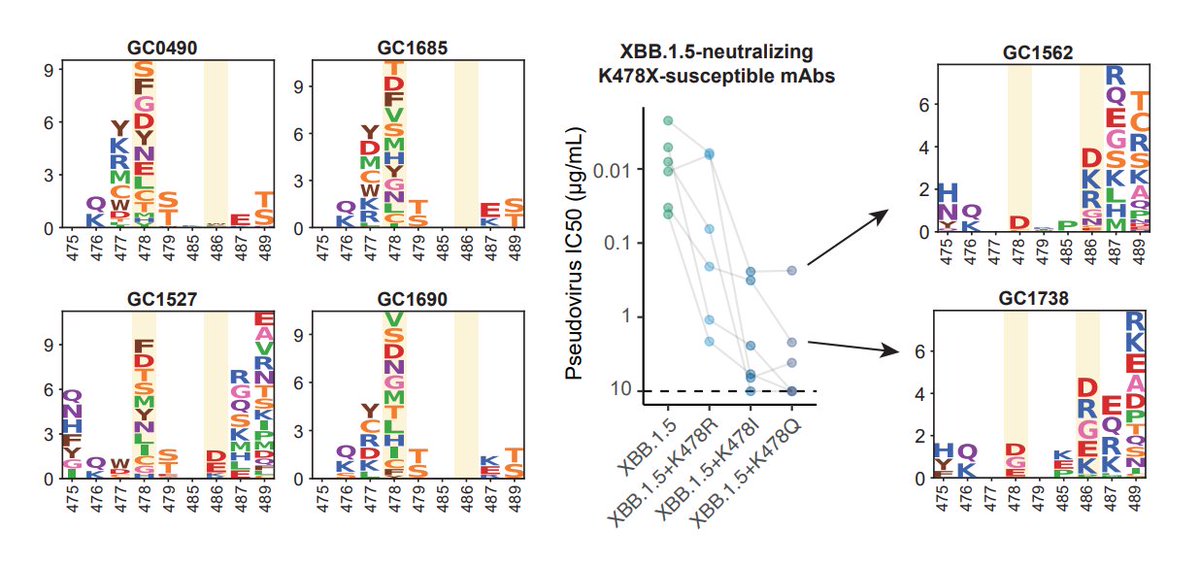
Another potential background that could introduce K478X is Delta-imprinted convalescents who experienced BA.5/BQ.1.1/XBB infections, which may allow the generation of abundant K478X-sensitive mAbs since Delta carries T478K. This might explain why K478X mostly appeared from India.
More detailed descriptions of the data will be shared next week on BioRxiv, together with new studies on immune imprinting dynamics and RBD evolution prediction of XBB*. A huge new update of DMS data for immune evasion and ACE2 binding prediction will also be shared. Stay tuned!
• • •
Missing some Tweet in this thread? You can try to
force a refresh