
In a recent preprint with @GorinGennady (biorxiv.org/content/10.110…) we provide a quantitative answer to to this question, namely what information about variance (among cells in a cell type, or more generally many cell types) does a UMAP provide? A short🧵1/
https://twitter.com/WallaceUcsf/status/1652772412776394753
The variability in gene expression across cells can be attributed to biological stochasticity and technical noise. In practice it's hard to break down the variance into these constituent parts. How do we know what is biological vs. technical? 2/
Here's an idea: within a cell type, we can obtain an accurate estimate of gene expression by averaging across cells. Now we can get a lower bound for biological variability by computing the variance across very distinct cell types. 3/
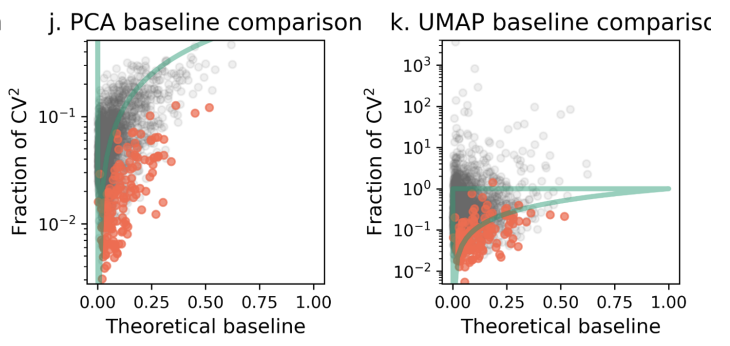
We can now assess whether a transformation of a count matrix is removing only technical noise, or throwing out some biological signal by accident. The bound means one should end up in the green region. Throwing out too much variance will place a gene outside of it. 4/ 

Consider the two first steps in every single-cell RNA-seq analysis:
1. Depth normalization (in the fig. below "PF" for proportional fitting).
2. Log-transform, i.e. log(x+1) of the data (in the fig. below "log")
Red points are the top highly expressed genes. 5/
1. Depth normalization (in the fig. below "PF" for proportional fitting).
2. Log-transform, i.e. log(x+1) of the data (in the fig. below "log")
Red points are the top highly expressed genes. 5/

What you can see is that depth normalization is ok. It adds a bit of noise (some genes have more variance than they started with) but not too bad. However the log transform drops many genes out of the admissible zone. That is, the transform has removed biological signal. 6/
The intuition behind variance stabilization, or normalization, namely that it's removing technical noise due to sampling etc., is not quite right. Yes, the transform is reducing technical noise, but it's throwing out some of the baby with the bathwater. 7/
Next the standard step is PCA. We see that even more biological signal is removed. The final step is UMAP, which adds tons of noise. Some of the points move back above the green line, but remember that biological signal has already been removed. It's just addition of noise. 8/
In other words, UMAP is the worst of all worlds. The procedure does not remove technical noise, it adds it in to the data. And for no reason: all one gets for noising the data is distortion of distances. 9/
https://twitter.com/lpachter/status/1431325969411821572?s=20
So what should one do instead? In our preprint we present Monod, software for *modeling* both of the biophysics and technical noise. This way, one doesn't have to throw out the baby with the bathwater, because one works to figure out the differences between them. 10/

Another advantage of Monod: it provides a natural way to "integrate" or "harmonize" different data modalities. It provides an answer as to which matrix to choose in an analysis: spliced or unspliced? Rather than choosing between them, with Monod they are used together. 11/

Furthermore, Monod generalizes differential expression testing to the identification of genes with distributional differences. We provide some interesting use cases, and also validate performance with respect to a recently published dataset from @Weinberger_Lab. 12/

Monod is available here: github.com/pachterlab/mon… 13/
Tutorials and examples are provided here: github.com/pachterlab/mon… 14/
Documentation is here: monod-examples.readthedocs.io/en/latest/ 15/
More on @GorinGennady related work in this thread:
Also a once-in-career opportunity to hire him: he is defending on May 19th. DM me for Zoom info if you're interested. 16/
https://twitter.com/lpachter/status/1635645224629448705
Also a once-in-career opportunity to hire him: he is defending on May 19th. DM me for Zoom info if you're interested. 16/
In summary, don't start your #scRNAseq analysis by throwing out almost all of the biological signal in your data. And to return to your question @WallaceUcsf, the UMAP for a single cell type is not much more than a Rorschach test. 17/17
• • •
Missing some Tweet in this thread? You can try to
force a refresh







