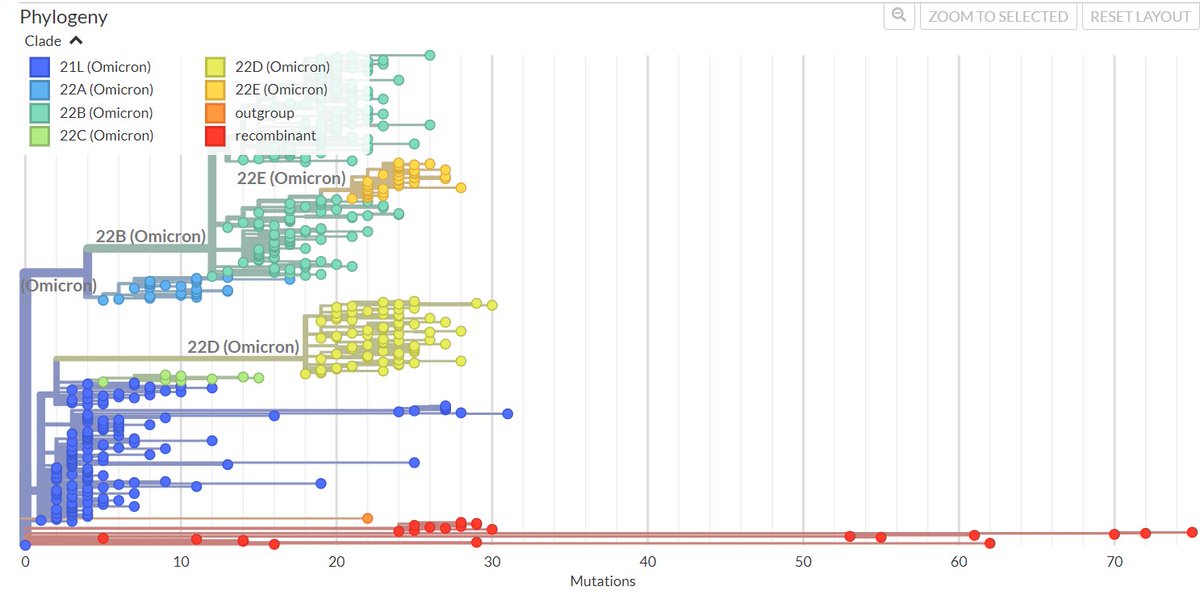
As BA.2.86 keeps on poppin, I'll outline key insights on its prevalence, genomic features, potential impacts, diagnostic advancements and highlight for areas requiring immediate focus.
For ongoing updates, please follow my page.
Let’s go:
For ongoing updates, please follow my page.
Let’s go:

Most important note :
Only 5 BA.2.86 cases verified so far by sequencing. that's not much.
We must analyze with caution to prevent overinterpretation. With such limited data, We can easily make more noise than data. Nevertheless, these few cases could signal an emerging concern, requiring continued vigilance and alertness.
Only 5 BA.2.86 cases verified so far by sequencing. that's not much.
We must analyze with caution to prevent overinterpretation. With such limited data, We can easily make more noise than data. Nevertheless, these few cases could signal an emerging concern, requiring continued vigilance and alertness.
The 5 cases spotted across 4 countries (US, UK, Denmark, and Israel). These cases were all identified within the community and occurred within a short time frame, specifically at the beginning of August. This geographical spread and rapid emergence warrant close monitoring and investigation

This is not Omicron.
Although it has evolved from one of Omicron's main branches (BA.2), it is as diverged from BA.2 as BA.2 was from the basal lineage of this pandemic.
Regardless of any official nomenclature that may continue to classify it as Omicron, if the case count reaches 20 samples across 10 countries by the end of next week, I propose referring to it as the Pi variant, emphasizing its unique identity.
Although it has evolved from one of Omicron's main branches (BA.2), it is as diverged from BA.2 as BA.2 was from the basal lineage of this pandemic.
Regardless of any official nomenclature that may continue to classify it as Omicron, if the case count reaches 20 samples across 10 countries by the end of next week, I propose referring to it as the Pi variant, emphasizing its unique identity.
Regardless of any inferences drawn from this extreme saltation, it's essential to recognize that the high number of mutations may complicate our ability to calculate or speculate based on known specific mutations. We must keep in mind the concept of epistasis in every analysis, acknowledging that the interaction of mutations can produce complex and unpredictable effects.
A new variant may lead to changes in virulence, transmissibility, and immune recognition. While everyone’s interest is of the alterations in virulence, it is important to recognize that this aspect will likely be the last to be determined if changes have indeed occurred (we’re talking probably several months in such cases). Ongoing monitoring and research are crucial to comprehensively assess the potential impact of this variant.
Immune evasiveness is a key concern with such a significant saltation in the S1 protein. @jbloom_lab has conducted vital calculations on this matter (link bellow), providing important insights. If your not following him yet, this is the time to start.
*However, as previously stated, it's essential to remember the complexity of epistasis, which may affect our understanding and interpretation of these findings.
*However, as previously stated, it's essential to remember the complexity of epistasis, which may affect our understanding and interpretation of these findings.
https://twitter.com/jbloom_lab/status/1691826218738536747?s=20
The transmissibility of this variant may be inferred from the fact that the 5 samples have a notably low genomic distance between them. This indicates that they diverged from their most common recent ancestor very close to the time they appeared. Aassuming the molecular clock has remained relatively constant in this variant, this means not more than 2-4 weeks.
Combining this with the simultaneous appearance in community infections, across 4 different countries on 3 continents, provides a hint of rapid transmission. Although it's very early to make definitive conclusions, if this trend continues in the next samples, the situation may become concerning.
Combining this with the simultaneous appearance in community infections, across 4 different countries on 3 continents, provides a hint of rapid transmission. Although it's very early to make definitive conclusions, if this trend continues in the next samples, the situation may become concerning.
Data is currently missing, and sequence prevalence has dramatically reduced in recent months. Throughout the pandemic, new variants were first detected in border diagnostics and then within the community; however, border checks been cancelled some time ago. Therefore, I urge all laboratories worldwide not to delay the release of results. Please upload data to @GISAID ASAP and avoid stacking samples. Even if it means sequencing on smaller batches for the next week or two, we need this data more than any time in the past year.
Diagnostics have been updated.
You can refer to the list I've compiled of ultra-specific mutations (which I assess will be less prone to problems and may also detect sibling saltating variants, as seen with BA.1 & BA.2, B.1.617.1 & B.1.6172, and others).
Nextclade has been updated and can accurately detect BA.2.86 samples. I've tested its performance with synthetically made samples from BA.2.86, even with extreme defects and added genomic changes, and found it to be accurate; please use it.
USHER has also been updated to distinguish BA.2.86.
So @CorneliusRoemer , @AngieSHinrichs @theosanderson and all ur colleagues, From myself and from public health diagnostic teams worldwide, thank u for ur quick response and amazing work through the pandemic and now specificaly.
You can refer to the list I've compiled of ultra-specific mutations (which I assess will be less prone to problems and may also detect sibling saltating variants, as seen with BA.1 & BA.2, B.1.617.1 & B.1.6172, and others).
Nextclade has been updated and can accurately detect BA.2.86 samples. I've tested its performance with synthetically made samples from BA.2.86, even with extreme defects and added genomic changes, and found it to be accurate; please use it.
USHER has also been updated to distinguish BA.2.86.
So @CorneliusRoemer , @AngieSHinrichs @theosanderson and all ur colleagues, From myself and from public health diagnostic teams worldwide, thank u for ur quick response and amazing work through the pandemic and now specificaly.
@jbloom_lab This is it for now. ill update more if data will accumulate, but hope it will not.
• • •
Missing some Tweet in this thread? You can try to
force a refresh