
 This analysis is based on @GISAID data (a resource that revolutionized how we track 🦠evolution).
This analysis is based on @GISAID data (a resource that revolutionized how we track 🦠evolution). Wild birds across the Americas (and globally) facing an avian flu panzootic in recent years, from a European origin. And as the load is high in birds, we see more and more spillovers, even to places hosts we wouldnt guess before.
Wild birds across the Americas (and globally) facing an avian flu panzootic in recent years, from a European origin. And as the load is high in birds, we see more and more spillovers, even to places hosts we wouldnt guess before.

 In this thread, I will review the different methods for taxonomy and nomenclature of flu lineages and strains, and provide my suggestion for the first step to create a standardized informative system.
In this thread, I will review the different methods for taxonomy and nomenclature of flu lineages and strains, and provide my suggestion for the first step to create a standardized informative system. We have three important factors in understanding why H5N1 in cattle will disappear:
We have three important factors in understanding why H5N1 in cattle will disappear: This tool is based on a method I previously presented, implementing the transition in SARS-CoV-2 to massive 🧬which allows for extensive coverage in many countries and enables setting a high standard for data quality.
This tool is based on a method I previously presented, implementing the transition in SARS-CoV-2 to massive 🧬which allows for extensive coverage in many countries and enables setting a high standard for data quality.  Genomic background: Dengue virus has 4 main serotypes, likely from different historical events. High diversity exists between and within serotypes. In the 1990s, lineages were defined using partial sequencing of the envelope protein (E).
Genomic background: Dengue virus has 4 main serotypes, likely from different historical events. High diversity exists between and within serotypes. In the 1990s, lineages were defined using partial sequencing of the envelope protein (E).
 So - There are two risks to address:
So - There are two risks to address:  First lets revisit the phylogenetics. Here is a RAxML build of the🧬for the HA segment.
First lets revisit the phylogenetics. Here is a RAxML build of the🧬for the HA segment.
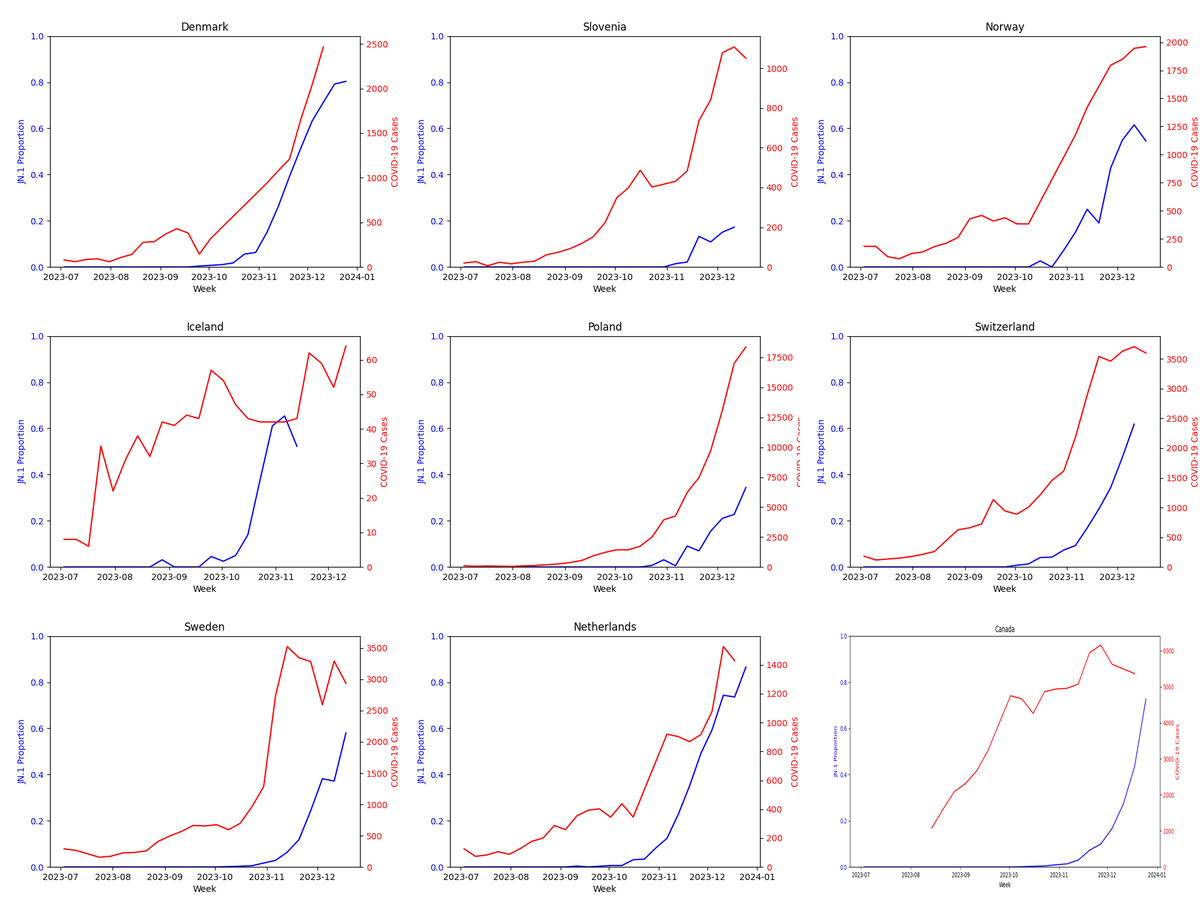
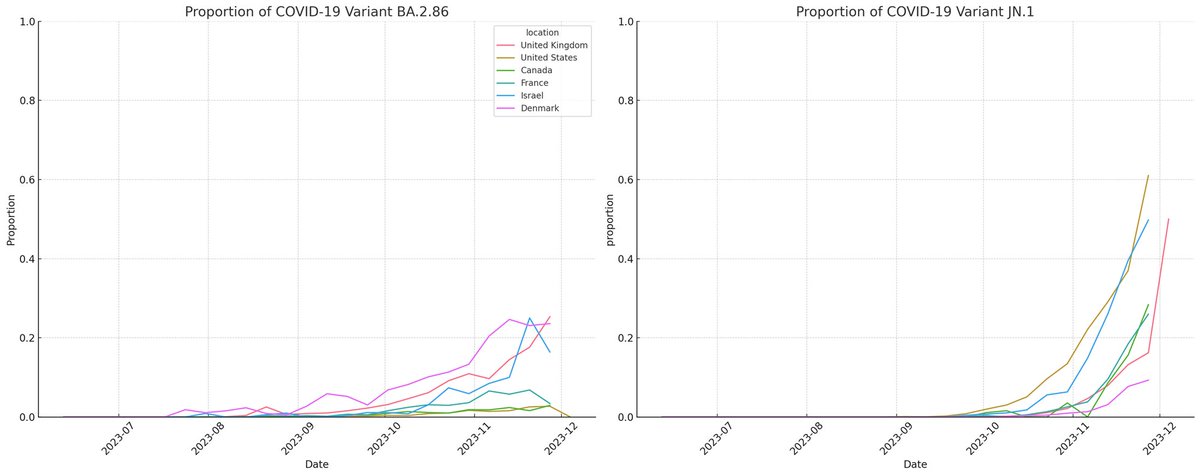
 Monitoring prolonged cases is key to predicting variant evolution and potential. Ideally, nations should focus on immunocompromised patients for this tracking and share their complete data on those. Without such targeted data, we'll have to make do with what's available.
Monitoring prolonged cases is key to predicting variant evolution and potential. Ideally, nations should focus on immunocompromised patients for this tracking and share their complete data on those. Without such targeted data, we'll have to make do with what's available. 1.
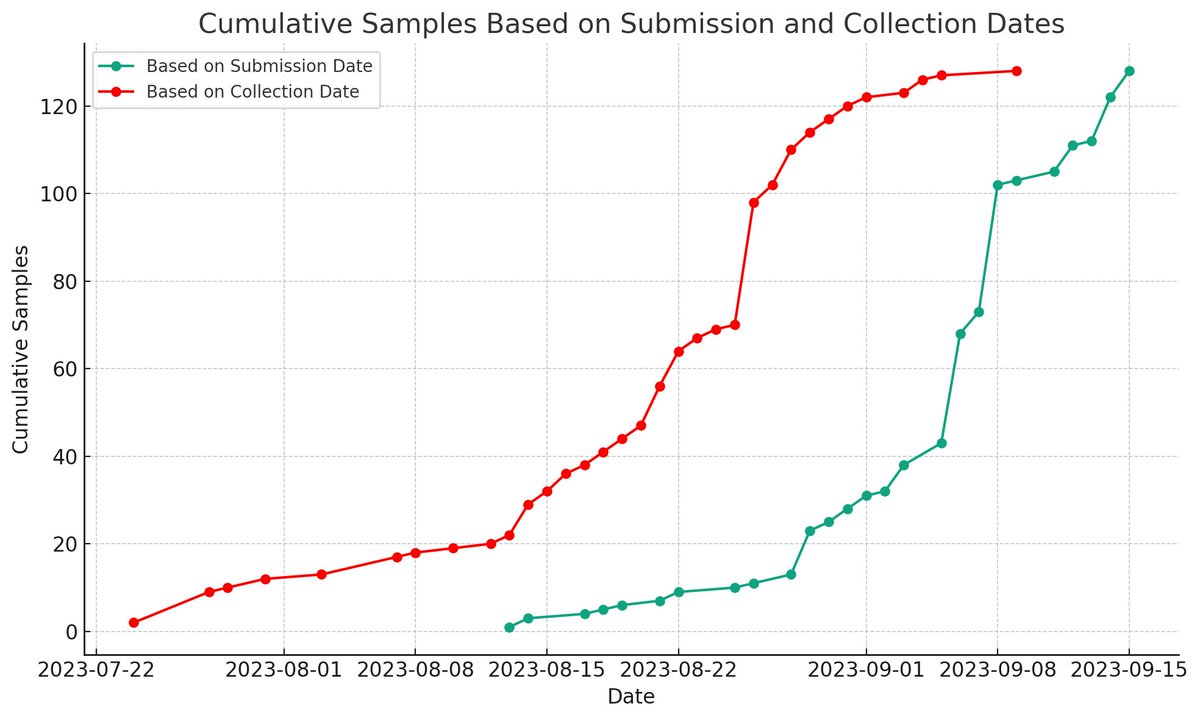
1.
 So lets start with the data.
So lets start with the data.  Two routes in the virus evolution:
Two routes in the virus evolution: Most important note :
Most important note : @GISAID If were taking it as a BA.2 2nd gen, it means it got :
@GISAID If were taking it as a BA.2 2nd gen, it means it got :
 I took the definitions of 526 Omicrons sub-variants (from a DB i maintain and will share in the end here). This entailed assessing the count of synonymous SNPs against RBD SNPs in each varaiant. Averaging # RBD SNP's for each # of synonymous SNP's.
I took the definitions of 526 Omicrons sub-variants (from a DB i maintain and will share in the end here). This entailed assessing the count of synonymous SNPs against RBD SNPs in each varaiant. Averaging # RBD SNP's for each # of synonymous SNP's. 
 Every entry goes through an analysis includes statistical examination of mutations, verification against USHER's Mutational path, study of parent's defining mutations, and analysis of mutation positions, including Ambiguous nucleotide and discontinuous regions.
Every entry goes through an analysis includes statistical examination of mutations, verification against USHER's Mutational path, study of parent's defining mutations, and analysis of mutation positions, including Ambiguous nucleotide and discontinuous regions.