Updates on BA.2.86.
1) BA.2.86's ACE2 binding affinity is very high.
2) BA.2.86 has lower fusogenicity than XBB.1.5.
3) BA.2.86's infectivity in Vero cells is similar to BA.1, lower than XBB.1.5.
4) Structure analysis shows that BA.2.86's Spike prefers RBD "down" conformation.
1) BA.2.86's ACE2 binding affinity is very high.
2) BA.2.86 has lower fusogenicity than XBB.1.5.
3) BA.2.86's infectivity in Vero cells is similar to BA.1, lower than XBB.1.5.
4) Structure analysis shows that BA.2.86's Spike prefers RBD "down" conformation.

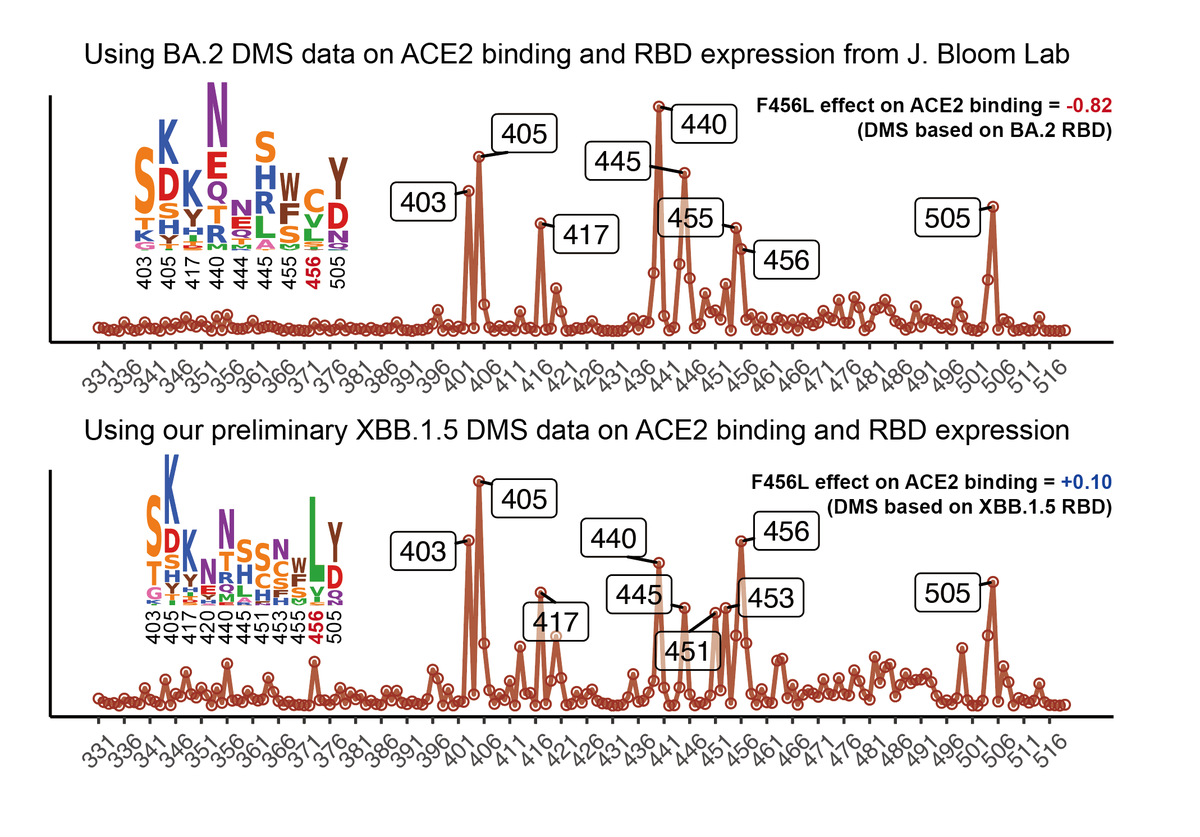
BA.2.86's RBD showed a pretty high hACE2 binding affinity measured by SPR, higher than that of XBB.1.5 and EG.5 and is even comparable to "FLip" variants like HK.3. BA.2.86's V483del indeed decreases ACE2 binding, but R403K is just too powerful and makes up for the loss. 2/n

We also measured the cell-cell fusion capability using Spike-transfected 293T cells and 293T-hACE2 cells. BA.2.86 showed a lower fusogenicity than XBB.1.5, despite the fact that BA.2.86's ACE2 binding affinity is much higher. Note this assay is free of pseudoviruses. 3/n

We also performed the pseudovirus infection assay in Vero cells and found BA.2.86's infectivity is still lower than XBB.1.5, and is comparable to BA.1. Since BA.1 can transmit pretty well, this suggests BA.2.86 can also prevail as long as its immune evasion is strong enough. 4/n

To investigate why BA.2.86 has higher ACE2 binding but lower infectivity and fusogenicity, Xiangxi Wang's lab helped us solved the cryo-EM structure of BA.2.86's spike. Surprisingly, most of BA.2.86's spike has its RBD in "down" position compared to XBB.1.5. 5/n

Favoring "down" RBDs may explain BA.2.86's lower fusogenicity and infectivity observed in vitro since only "up" RBDs can bind to ACE2. It also suggests that BA.2.86's mutation on SD1-SD2 (where D614G is located) may have affected its spike conformation or spike flexibility. 6/n

Indeed, BA.2.86's P621S mutation actually created a alpha-helix near the 630 loop, which could enhance the local stability. Also, BA.2.86's E554K mutation formed a salt bridge with 583E, which could also increase stability. These changes may greatly affect spike flexibility. 7/n

Some other interesting features of BA.2.86's spike include the additional N354 Glycan introduced by K356T mutation (expected), and that the V483 deletion does not disrupt the C488-C480 disulfate bond, which is the reason why BA.2.86's RBD can still bind well to ACE2. 8/n

In sum, we showed that BA.2.86 exhibits high ACE2 binding, but probably still suffers from lower fusogenicity and infectivity (pseudovirus). The reason is likely due to BA.2.86's mutation on SD1-SD2 (E554K and P621S), which may cause BA.2.86's spike to favor "down" RBDs. 9/n
Various factors can determine a virus's transmission, including immune evasion, ACE2 binding, fusogenicity, etc. Since BA.1 can dominate given its reduced in vitro infectivity (proven in many studies), BA.2.86 can also prevail if it can break through immunity as good as BA.1 did.
So, can BA.2.86 break through current immunity as efficiently as BA.1 did before? Multiple BA.2.86 studies have suggested the opposite. In my opinion, the current form of BA.2.86 can probably beat XBB.1.5, maybe EG.5 as well, but not "FLip" variants like HK.3. 11/n
However, BA.2.86 is evolving (BA.2.86.1 already assigned), and that's why we need to closely monitor its evolution, to see whether BA.2.86 can gain mutations or reversions (especially on Spike) that could increase its transmissibility or immune evasion. 12/n
• • •
Missing some Tweet in this thread? You can try to
force a refresh