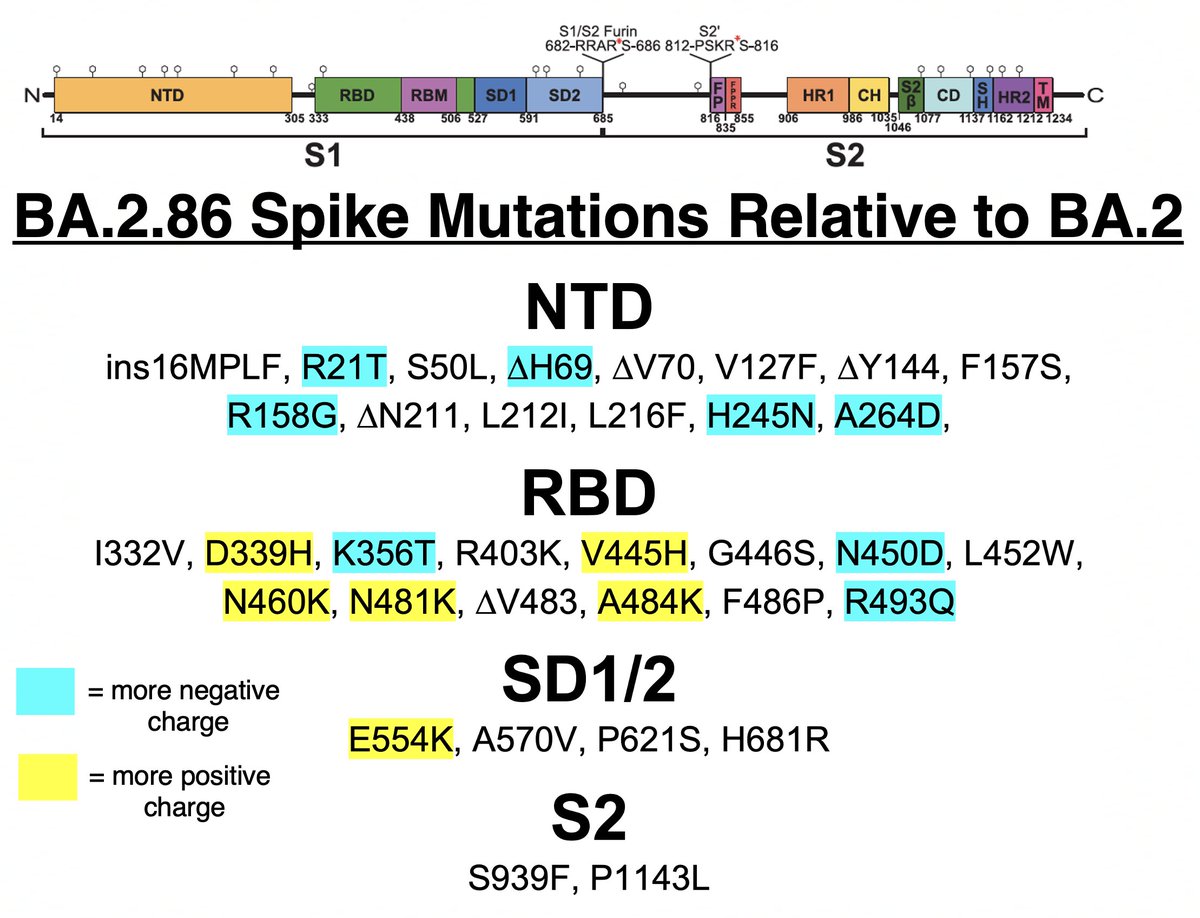
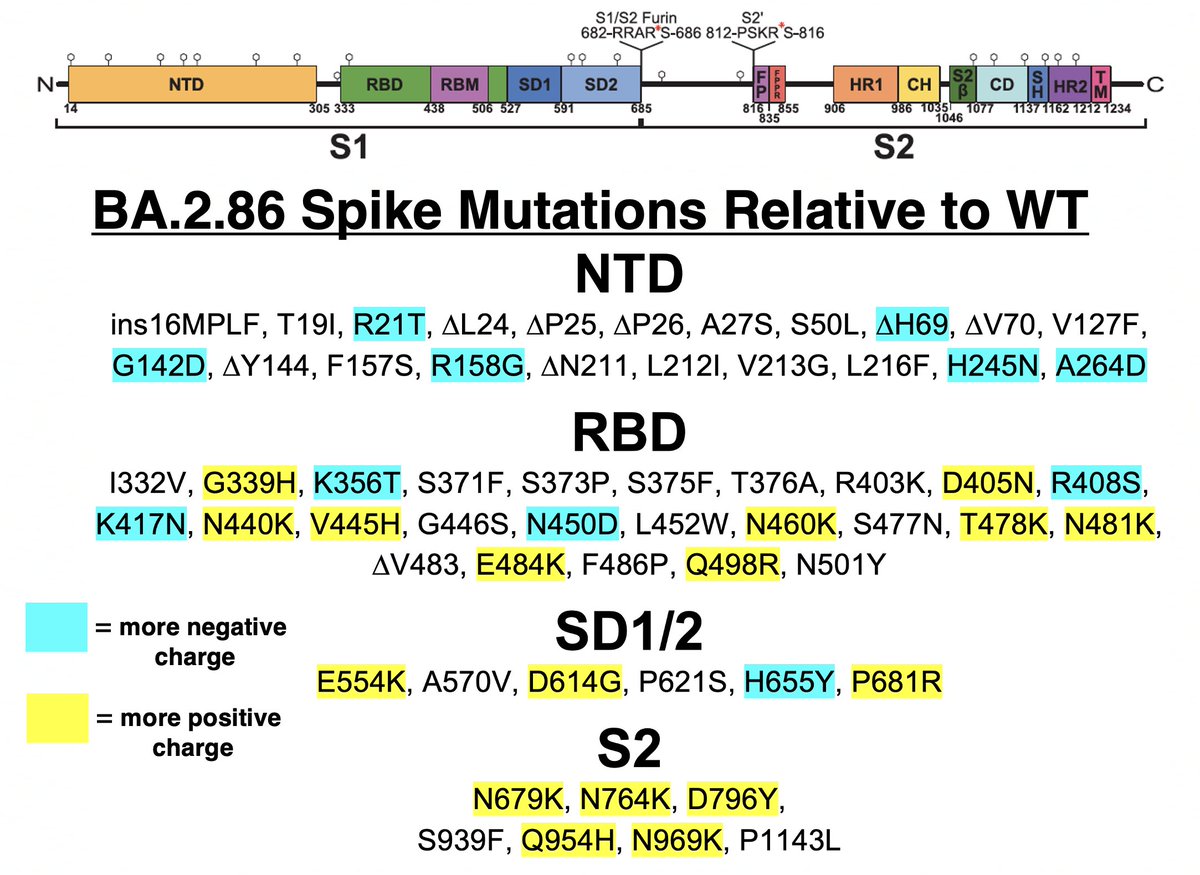
TL;DR—Among mutations common in chronic infections are two in PLpro, which combats innate immunity. Both mutations change the SARS-2 to a SARS-1-type AA & have clear effects—remarkable intrahost functional convergence. Then questions + wild speculation at thread's end. 1/58
If you follow SARS-CoV-2 evolution, you notice certain mutations patterns repeatedly appearing. This is even more true for chronic-infection sequences. The hallmark mutations pop up over & over. In some cases, we even have a decent idea of what they do. 2/
https://twitter.com/EricTopol/status/1697625461579477249
Thanks to the amazing work of @jbloom_lab & @tylernstarr, we can inspect almost any mutation in the spike RBD (receptor binding domain) and get a very good idea of how it affects ACE2 binding & spike stability. 3/58 tstarrlab.github.io/SARS-CoV-2-RBD…
And w/the spike antibody-escape calculator—supplemented by the rapid-fire updates of @yunlong_cao—we can see which spike mutations evade antibodies. Combined, these 3 measurements—ACE2 affinity, spike stability, & Ab evasion—have proven… 4/58
https://twitter.com/jbloom_lab/status/1658595410498437120
…astonishingly predictive. But more importantly, they are *explanatory.* They've provided a framework for interpreting how a given spike mutation functions & why it might appear & grow.
And coming to understand, even partially, one tiny corner of the world—that's pure gold. 5/
And coming to understand, even partially, one tiny corner of the world—that's pure gold. 5/
But the RBD is a small part of the spike protein, the rest of which remains much more enigmatic. Why, for example, are the Omicron S2 mutations—N679K, N764K, D796Y, Q954H, N969K—so implacably locked in, & what exactly do they do? 6/58 
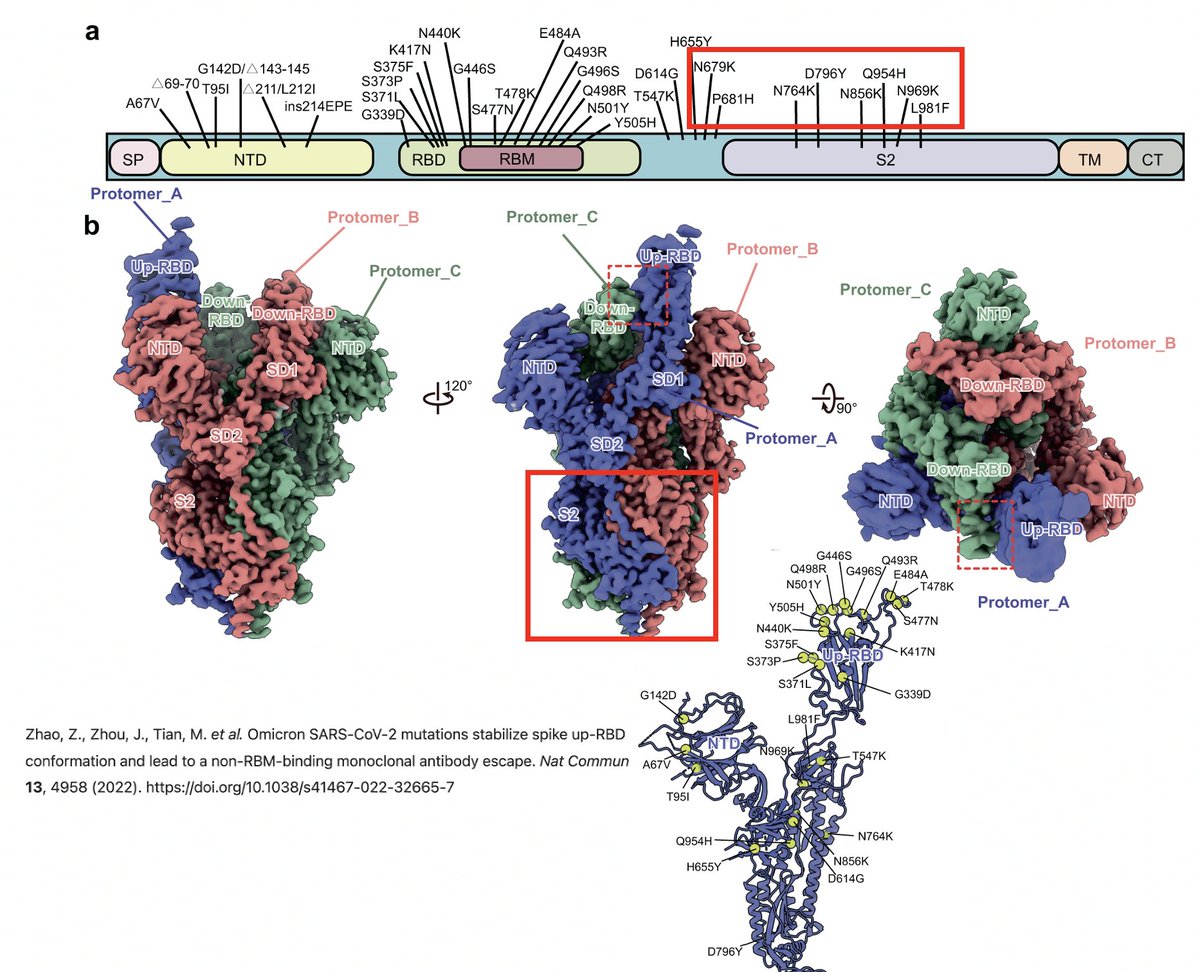
The function of some changes in the spike NTD—such as its recurrent deletions—we have some idea of, thanks to superb investigative work by @GuptaR_lab, @GroveLab, @veeslerlab, @mccarthy_kr, & others. Previous thread on this topic: 7/58
But what about the rest of the SARS-CoV-2 genome? As @shay_fleishon reminds us, the other 90% of the genome isn't there for decoration. If you see certain non-spike mutations happening again and again, you can bet there's a reason for it. 8/58
https://twitter.com/shay_fleishon/status/1472198436397195267
But discerning the effect a given mutation has, the reason it frequently appears, is usually impossible. Sometimes, we only have a vague idea about what a given genomic region does, or even no idea at all—see the saga of the s2m stem. 9/58
https://twitter.com/SolidEvidence/status/1590072665561497601
Most mysterious of all are the non-spike mutations that are rarely or never seen in major circulating lineages but which repeatedly sprout, like some exotic desert weed, from the infertile soil of chronic infections & cryptic wastewater lineages. 10/58
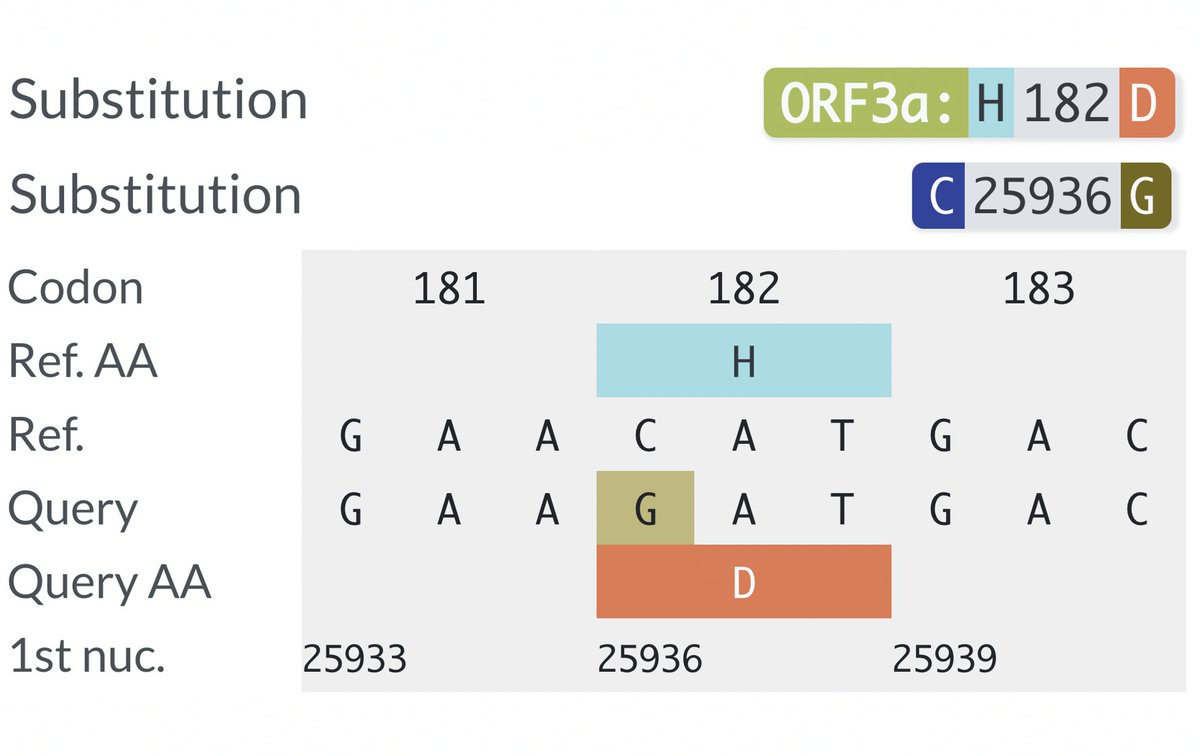
There are dozens of such chronic-convergent mutations, but in terms of frequency, two stand above the rest. Both are in the papain-like protease (PLpro), which is part of NSP3. First is NSP3_K977Q, also known as ORF1a:K1795Q. Thanks @ThornLab for image. 11/58
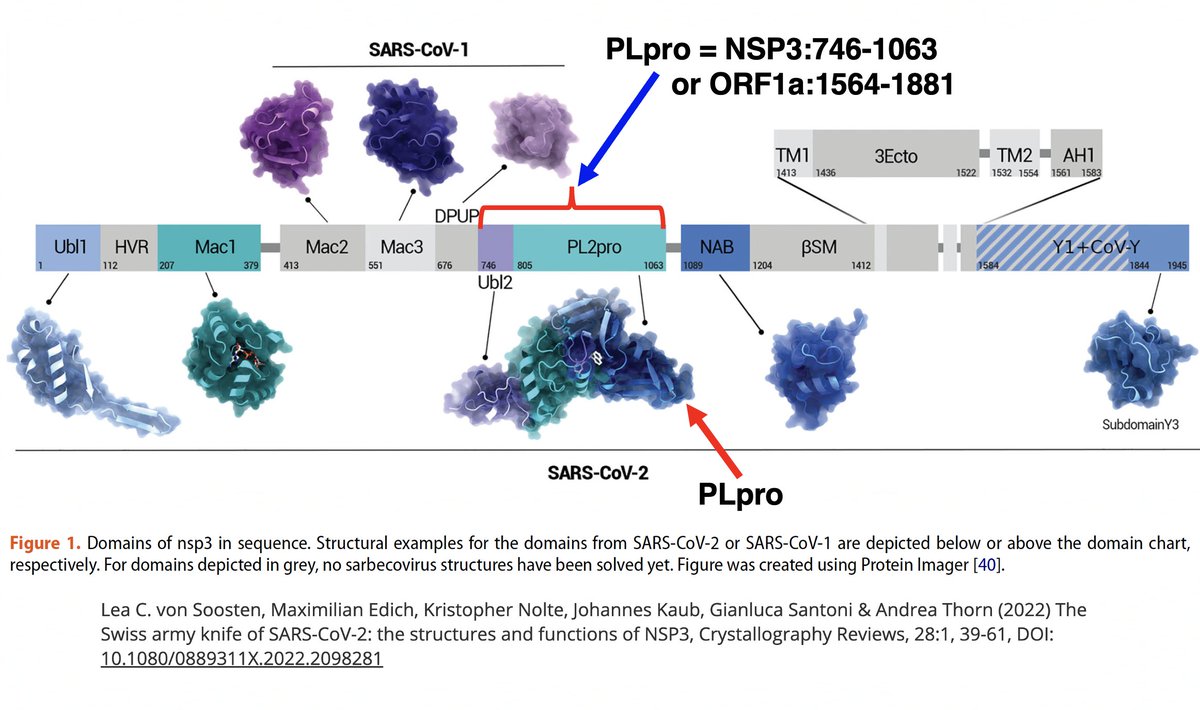
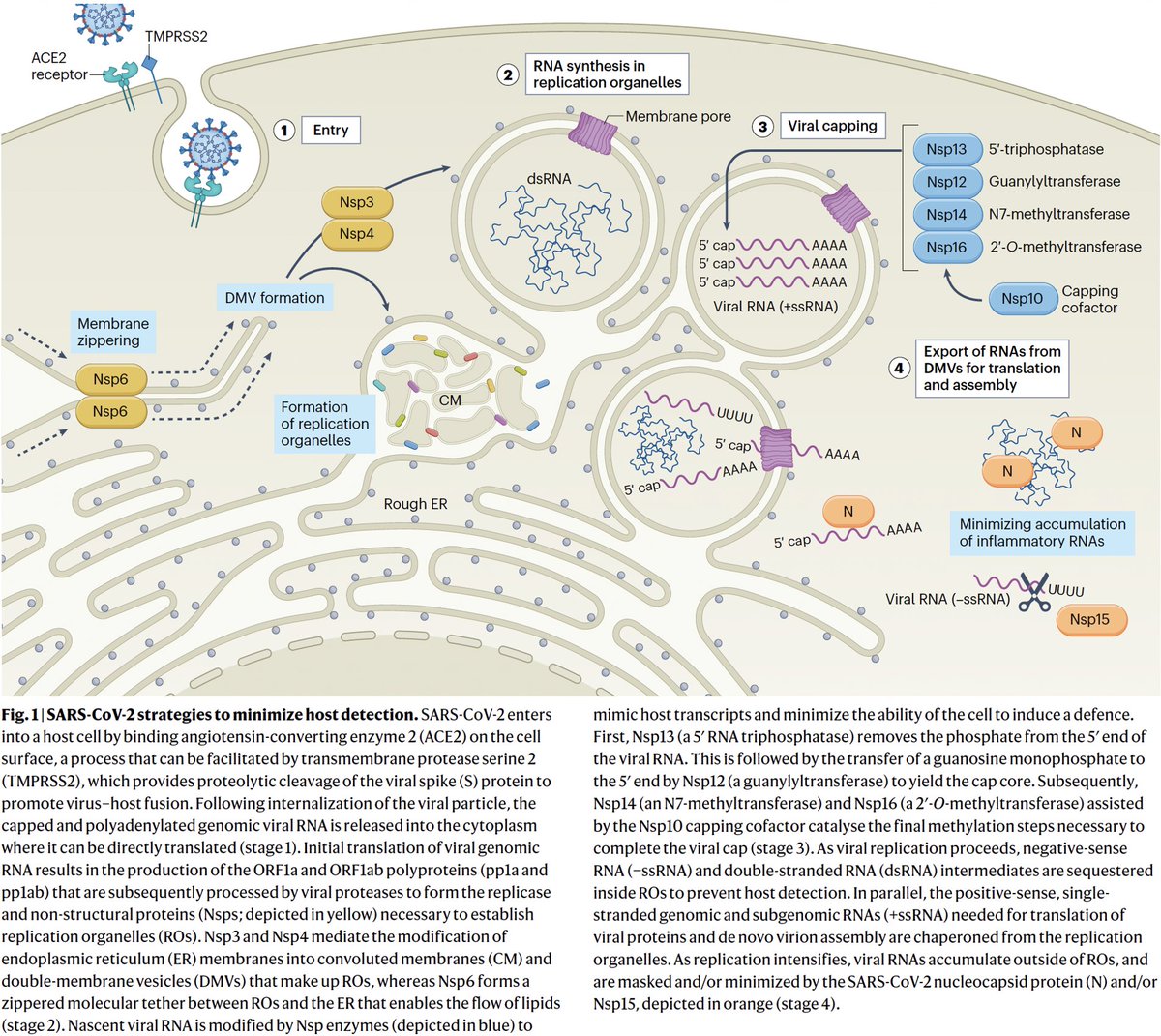
What is PLpro, and what does it do? That requires some brief background. When the full-length SARS2 genome is fed into a ribosome, first 70% or so—termed ORF1ab—is translated into an enormous string of amino acids (AAs). 12/58
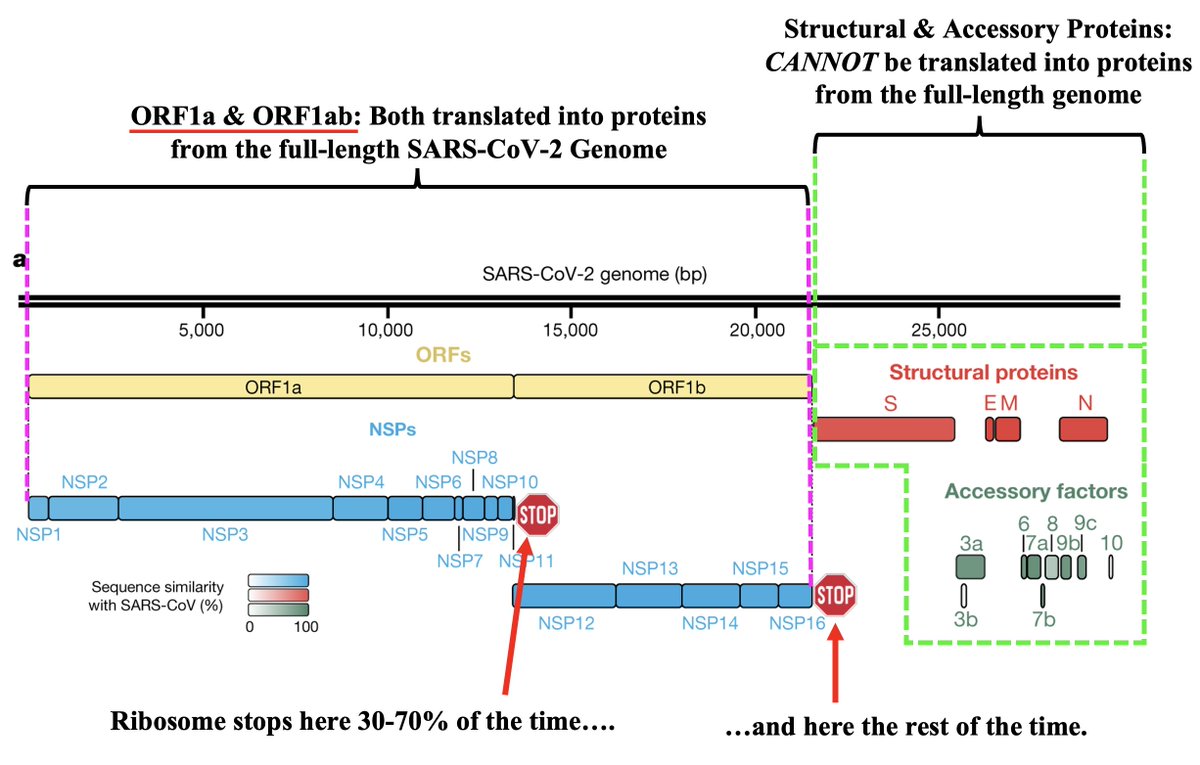
But this ORF1ab string (termed the ORF1ab polyprotein) is useless. To function, it has to be chopped into 15 separate proteins called non-structural proteins, NSP1 to NSP16 (skipping NSP11). Collectively, NSPs 1-16 are sometimes called the replicase because… 13/58
… they contain all the essential elements for genome replication. PLPro's primary function is to make the first three cuts to the ORF1ab polyprotein, freeing NSP1, NSP2, and NSP3. PLPro is located in NSP3, so it frees itself. Good video illustrating this by @MaastrichtU 14/58
The other 12 ORF1ab proteins are cut free by NSP5, also called the main protease (Mpro). Mpro is the target of Paxlovid. It's a very conserved protein, & contrary to some lurid headlines, there are no signs of serious Paxlovid resistance. 15/58
https://twitter.com/LongDesertTrain/status/1641436476079759361
NSP3, the largest protein in SARS-CoV-2, functions in countless ways—one paper calls it "The Swiss army knife of SARS-CoV-2"—and can divided into roughly 16 domains based on structure & function. PLpro is one of these domains. 16/58
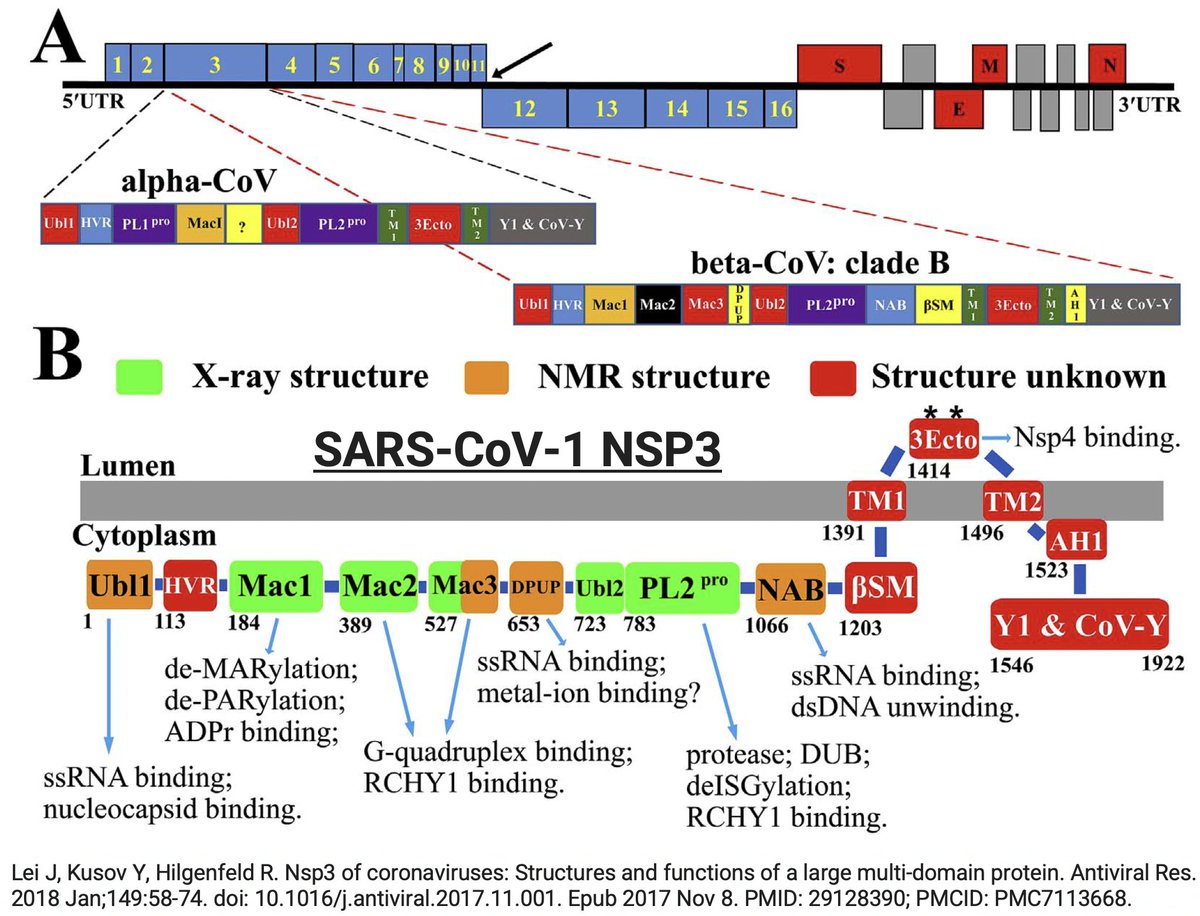
Many of the best sources of info on the various NSP3 domains are based on SARS-CoV-1, which is genetically similar to SARS-CoV-2. Very little is known about some of these domains—see e.g. some excerpts below from a superb review of NSP3 from 2018. 17/58

Even after dividing NSP3 into 16 domains, some domains have multiple functions. This is a constant theme in virology: To conserve genome size, proteins usually multi-task. See innate immune evasion functions alone—from superb review by @JudyMinkoff & @virusninja. 18/58
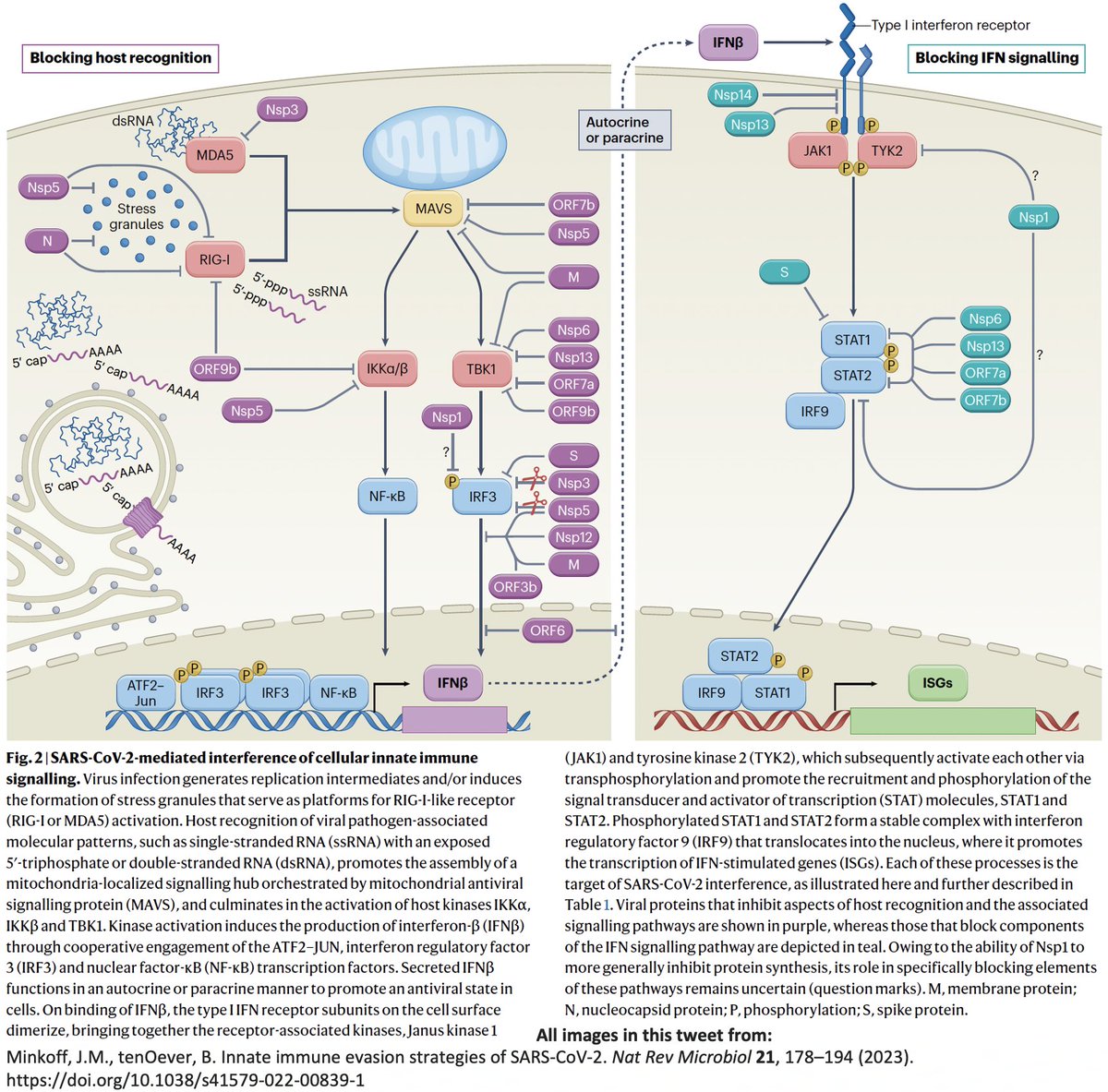
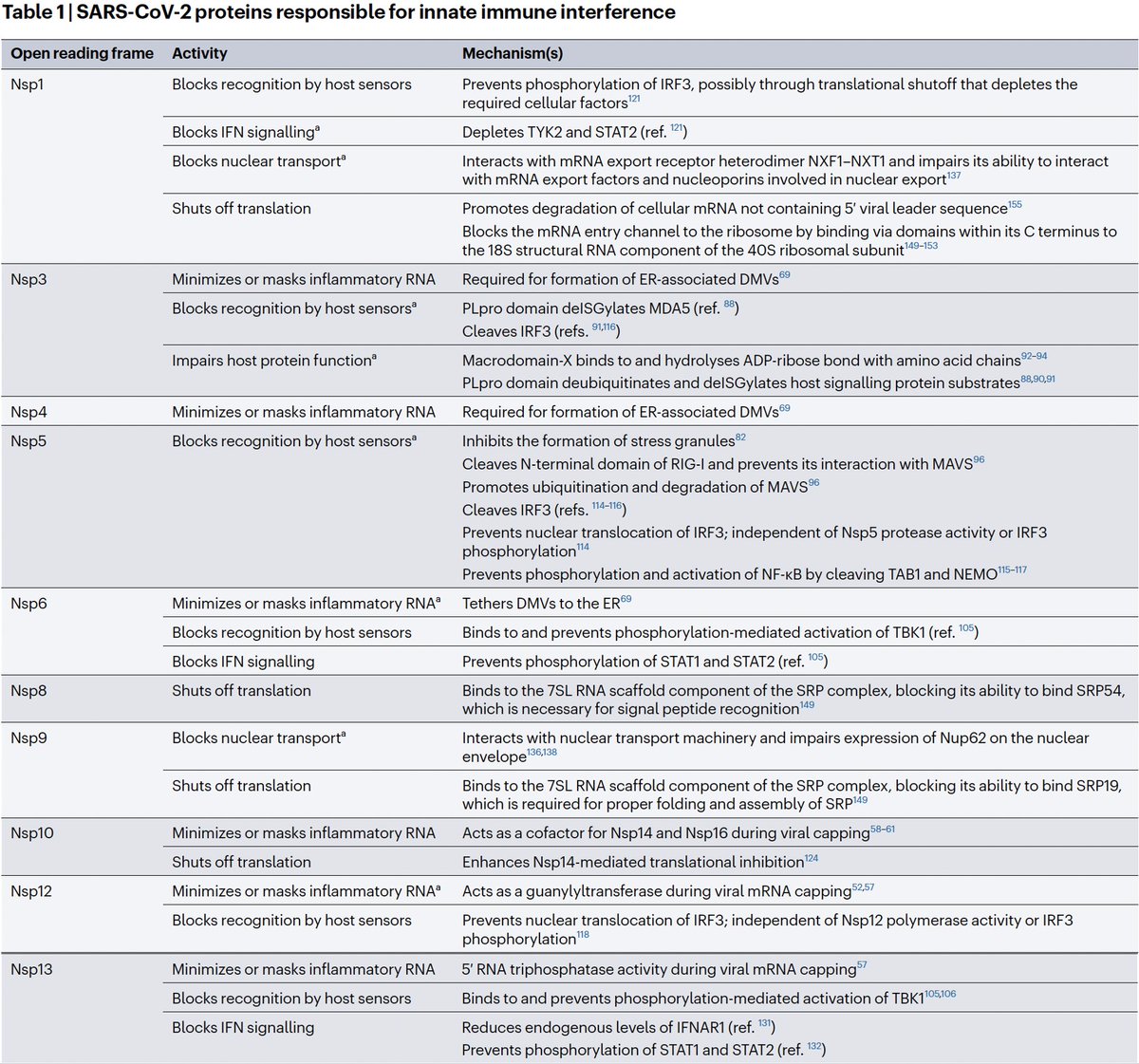
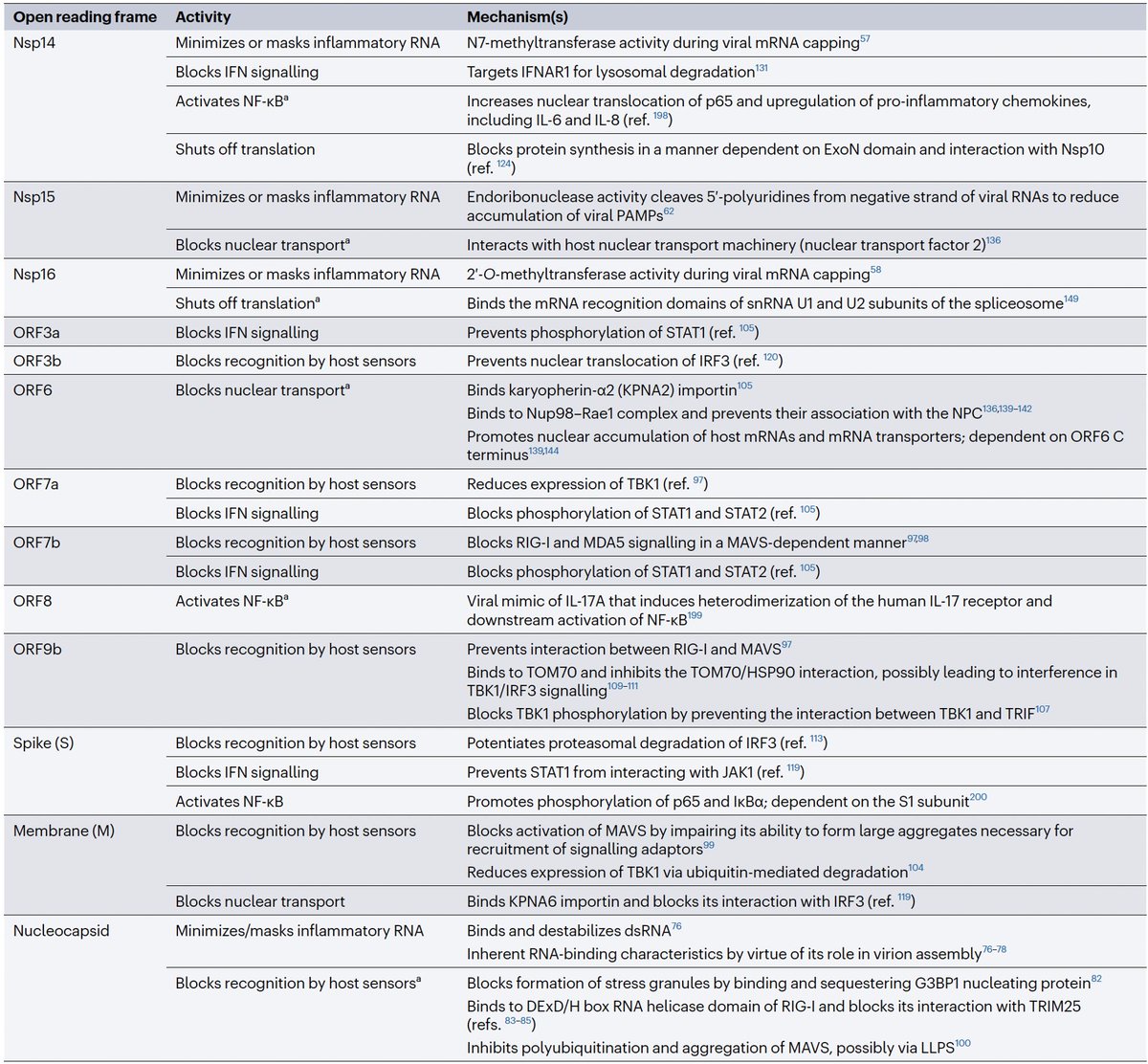
PLpro is no exception. Due to its vital role in replication & immune evasion (discussed below), it is probably the most well-studied part of NSP3. Researchers are seeking drugs to target PLpro, as you can see by the PubMed search results for "Papain" & "SARS-CoV-2." 19/58
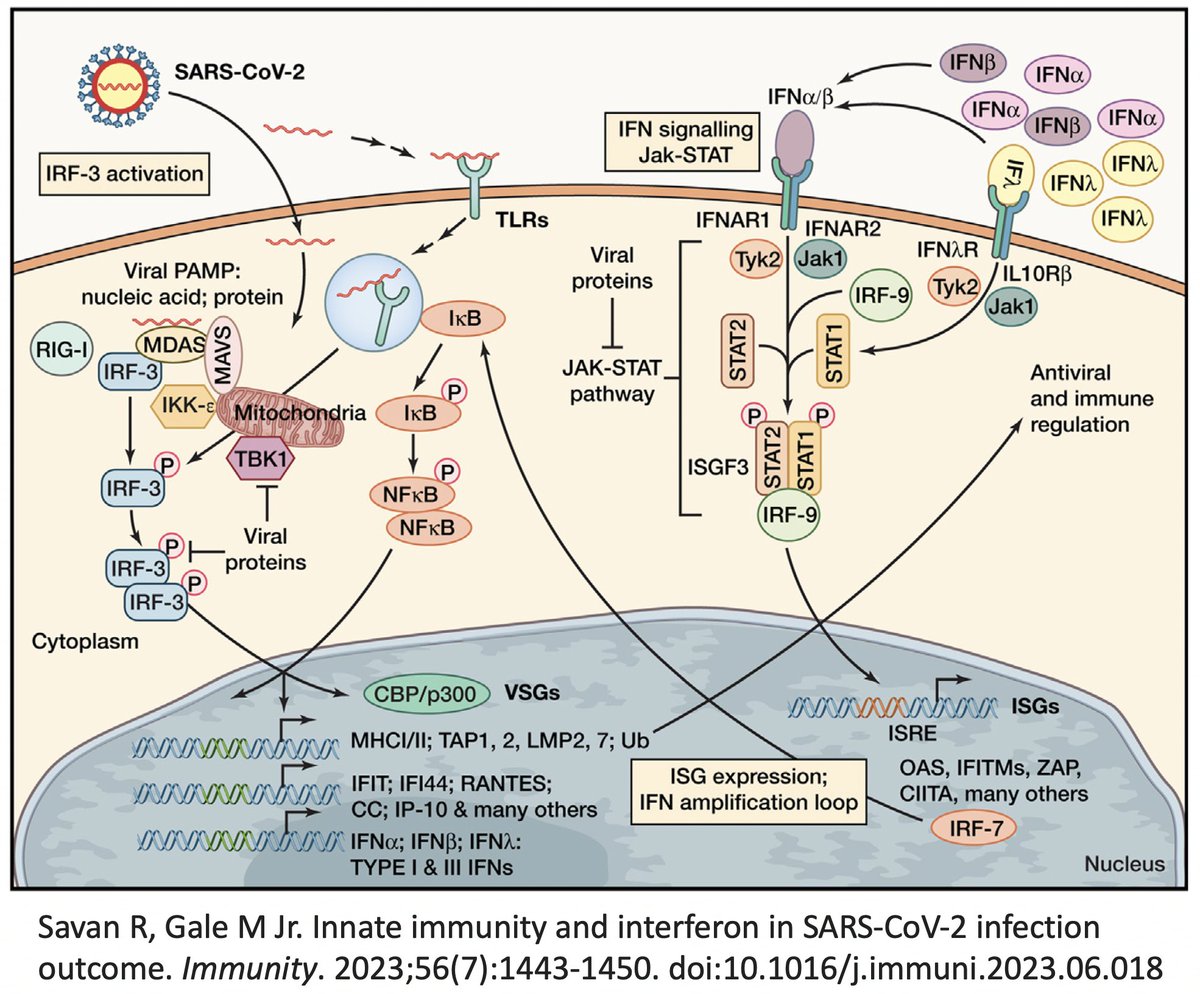
In addition to its essential role in replication, PLpro also abets coronaviruses' ability to evade the innate immune system, particularly the crucial interferon (IFN) response, which involves activation of hundreds of antiviral genes, called IFN-stimulated genes (ISGs). 20/58
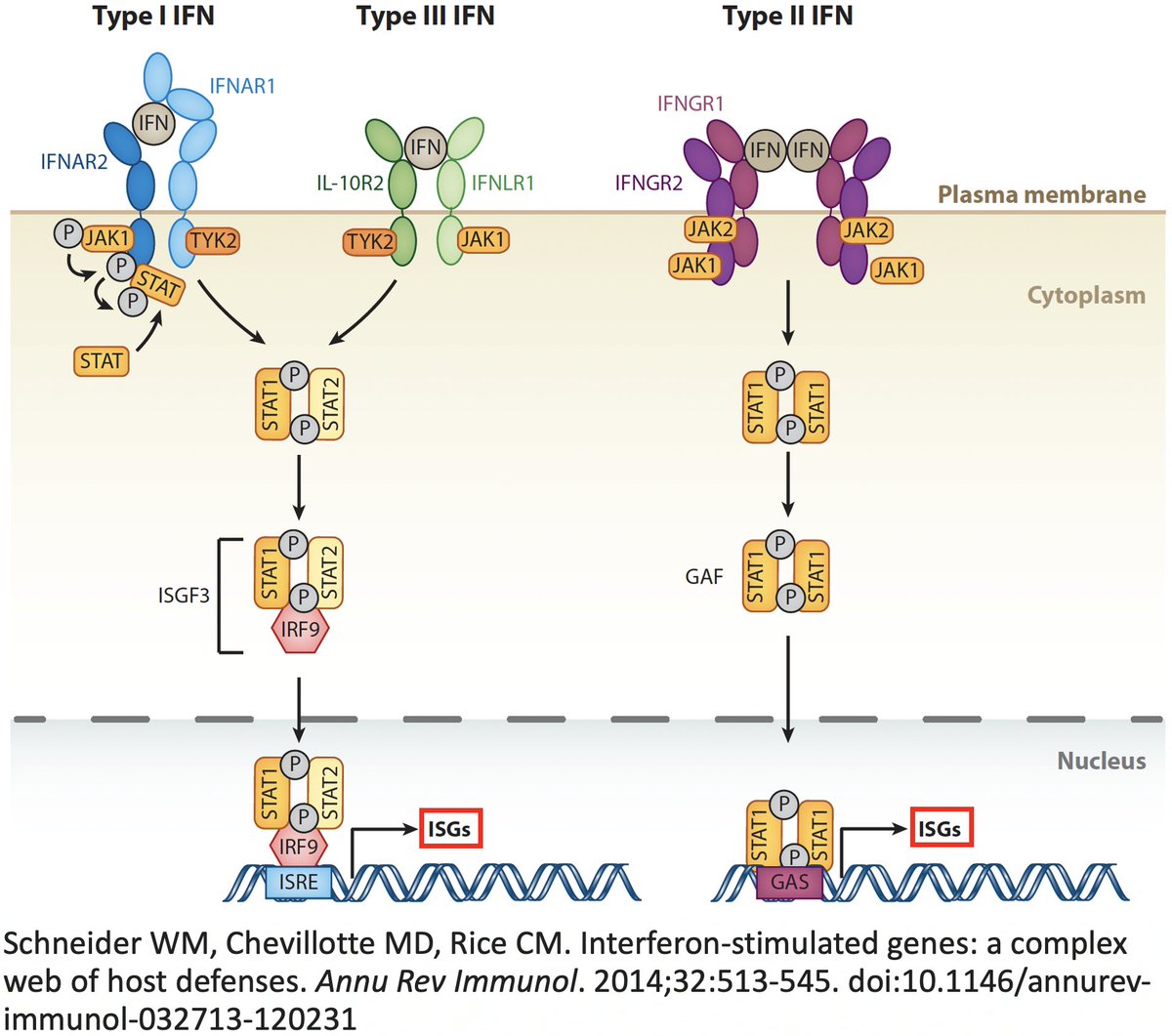
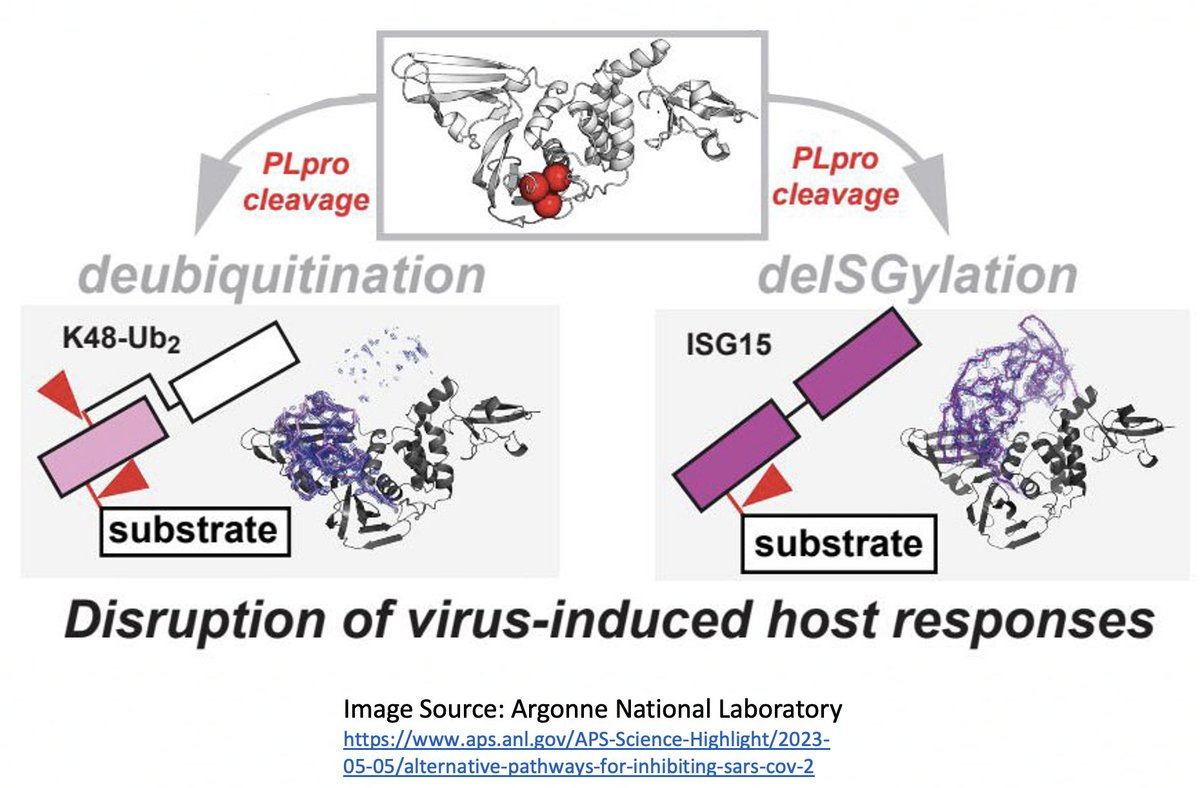
Here I'll focus on two known methods the SARS-CoV-2 PLpro employs to evade innate immunity: deubiquitination (DUB) and deISGylation (deISG) activity, both explained below—specifically, K48-linked DUB and ISG15 deISG. 21/58
Briefly, ubiquitin (Ub) is a small protein that gets attached ("conjugated") to a multitude of host and viral proteins, typically in the form of polyUb chains. This affects their function & fate in complex & diverse ways. This attachment process is called "ubiquitination." 22/58
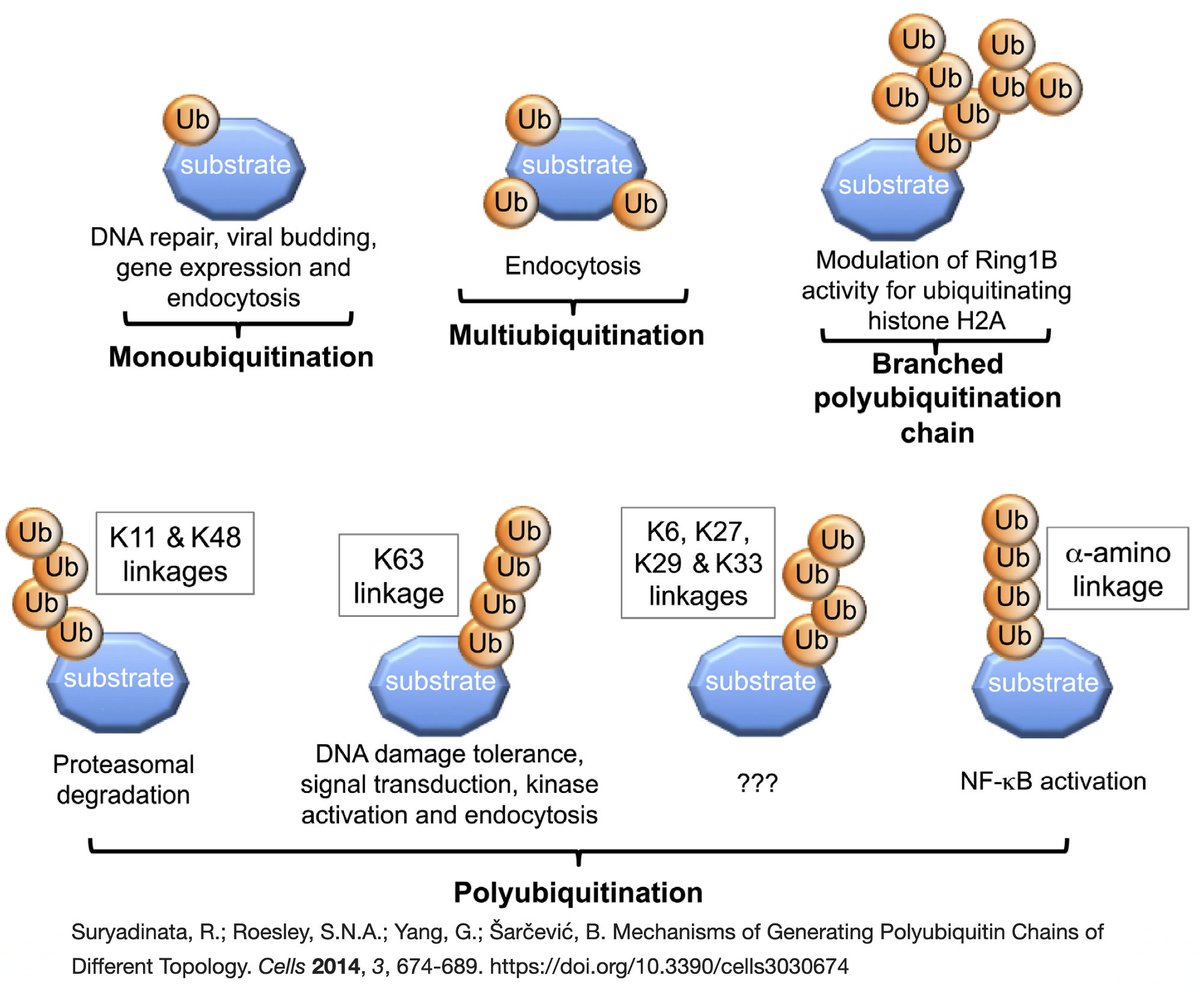
K48 ubiquitination usually marks a protein for destruction by the proteasome, a sort of predatory cytosolic sea cucumber that wanders the cytoplasm, devouring anything redolent of ubiquitin. Most often, ubiquitination dampens an immune response. Two examples… 23/58
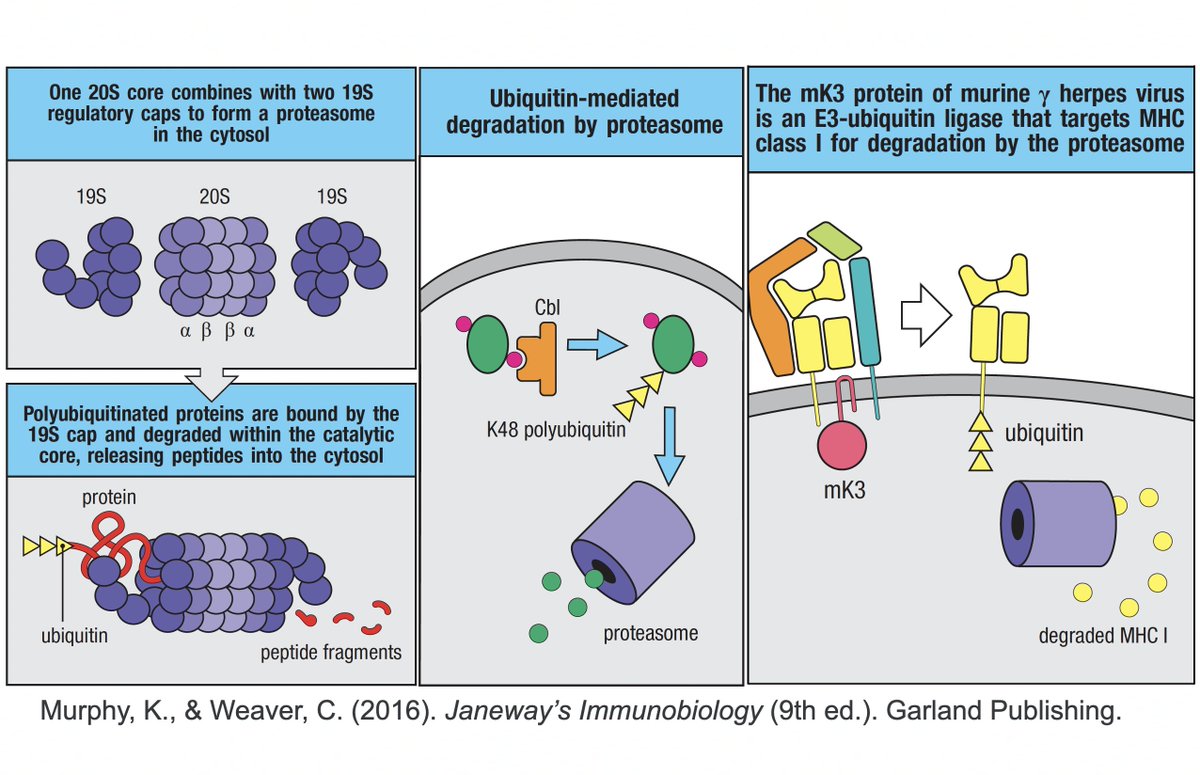
• Ubiquitination of the viral immune sensor & immune activator RIG-I result in its degradation, reducing the interferon response.
• Ubiquitination of MAVS, an IFN-and-inflammation-activating protein, marks it for proteasomal annihilation, tempering the immune response. 24/58
• Ubiquitination of MAVS, an IFN-and-inflammation-activating protein, marks it for proteasomal annihilation, tempering the immune response. 24/58


ISG15 is a protein made by one of many ISGs vital to our antiviral innate immune defense. Structurally, ISG15 resembles Ub, and like Ub, it gets attached to myriad proteins—host & viral—altering their activity. Both Ub & ISG15 play crucial roles in immune regulation. 25/58
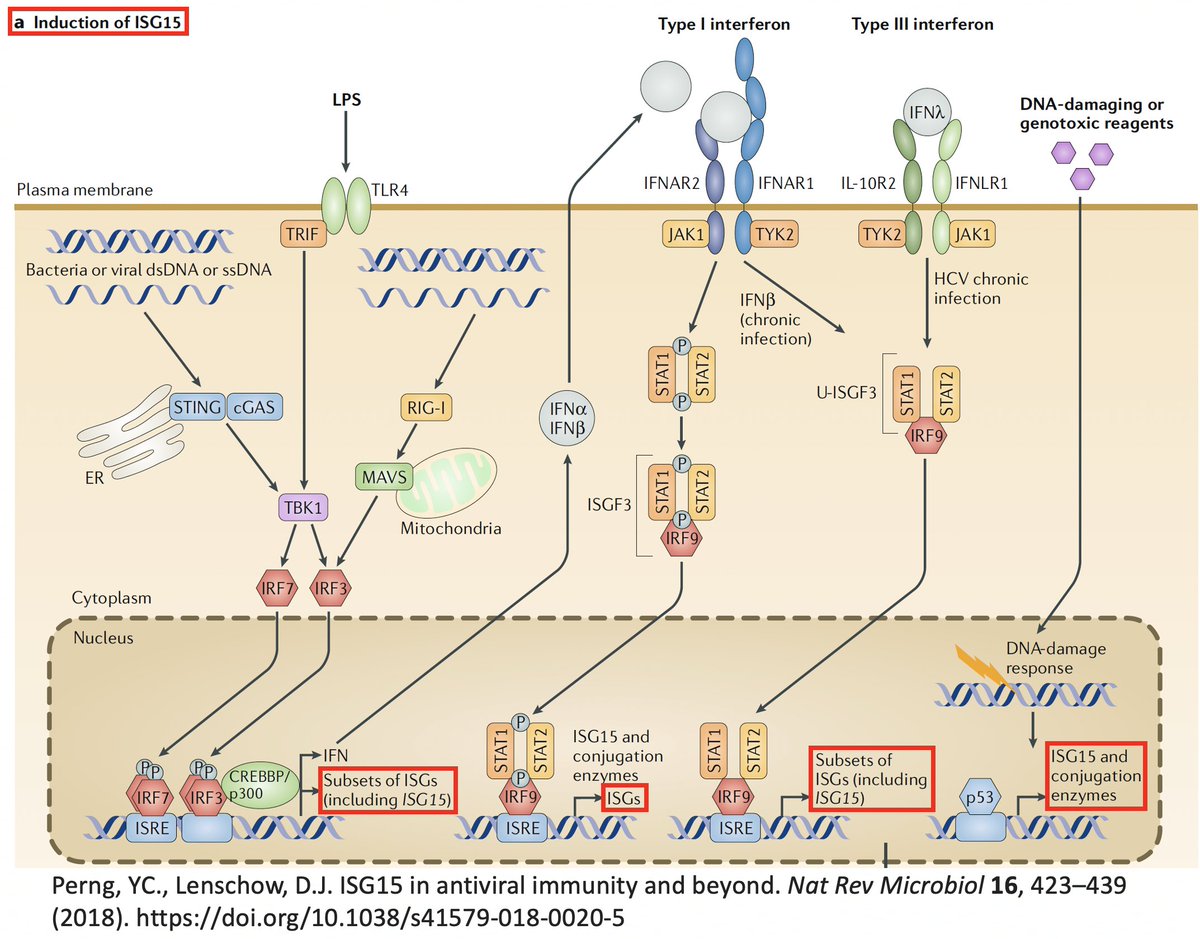
ISG15 usually activates an immune response. E.g.
• Virus: ISGylation of influenza B nucleoprotein (NP) prevents them from joining, preventing virion formation.
• Host: ISGylation of proteins involved in cellular trafficking prevents HIV budding & release. 26/58
• Virus: ISGylation of influenza B nucleoprotein (NP) prevents them from joining, preventing virion formation.
• Host: ISGylation of proteins involved in cellular trafficking prevents HIV budding & release. 26/58
As w/anything involving the immune system, matters are unspeakably convoluted. So in addition to activating innate immune mechanisms, ISG15 can also downregulate the immune response. Similarly, K48 ubiquitination not only dampens but also activates immune responses. 27/58


Given this complexity, how can we even know if ubiquitination & ISGylation combat viral infection? First, both are upregulated upon viral infection. This is not an accident. Second, viruses have devised numerous ways to combat both processes—also not an accident. 28/58

For our purposes, what's important is that all coronavirus PLpros *remove* both ubiquitin and ISG15 that are attached to viral & host proteins. Again, these processes are called, naturally enough, deubiquitination (DUB) and deISGylation (deISG). 29/58
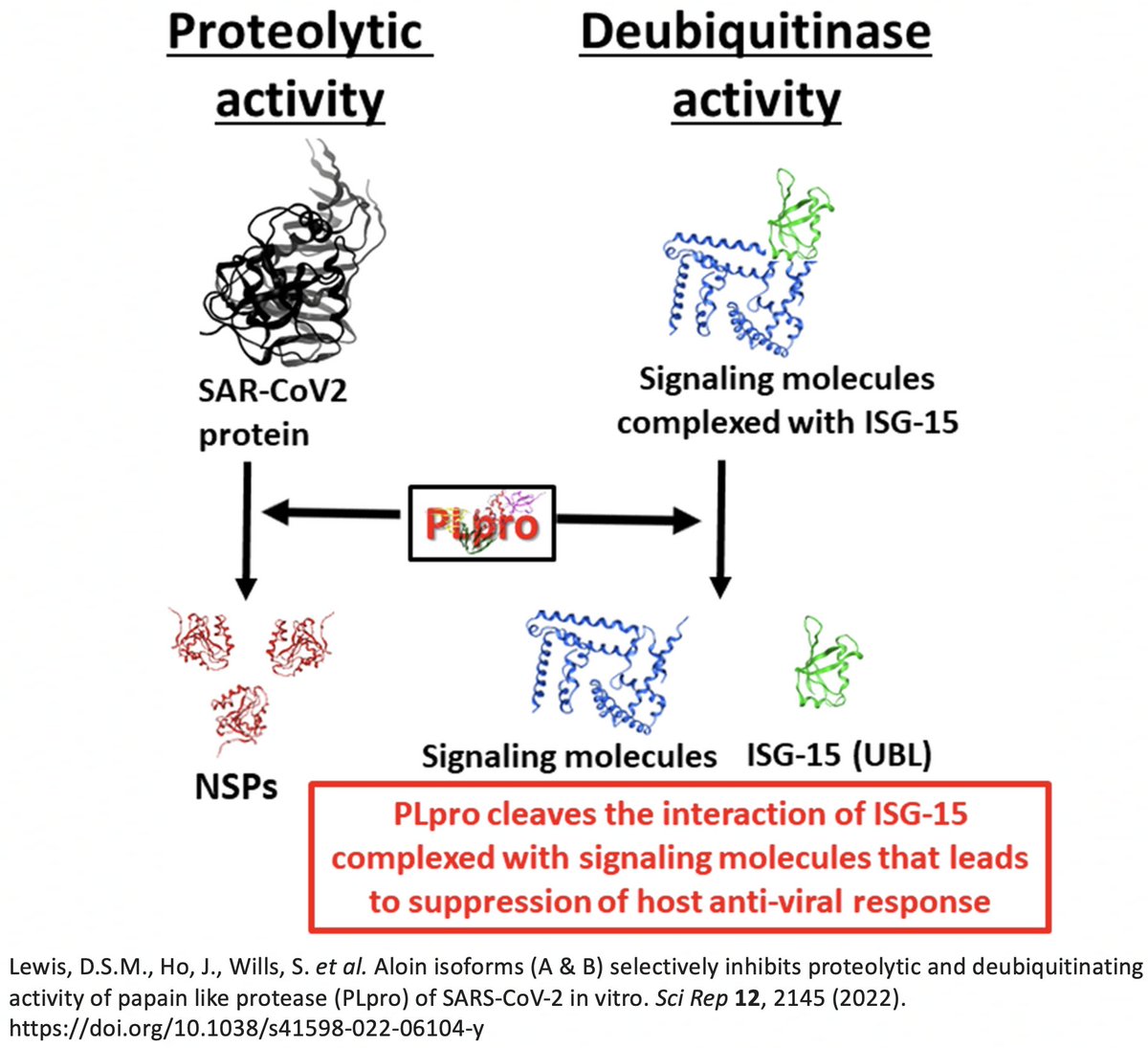
While the SARS-2 & SARS-1 PLpros are equally good at deISG, SARS-2 is far less efficient at DUB. Two superb studies analyzed the structure & sequence of the SARS1 & SARS2 PLpros & sought to determine why SARS1 so easily outperforms SARS2 at DUB. 30/58
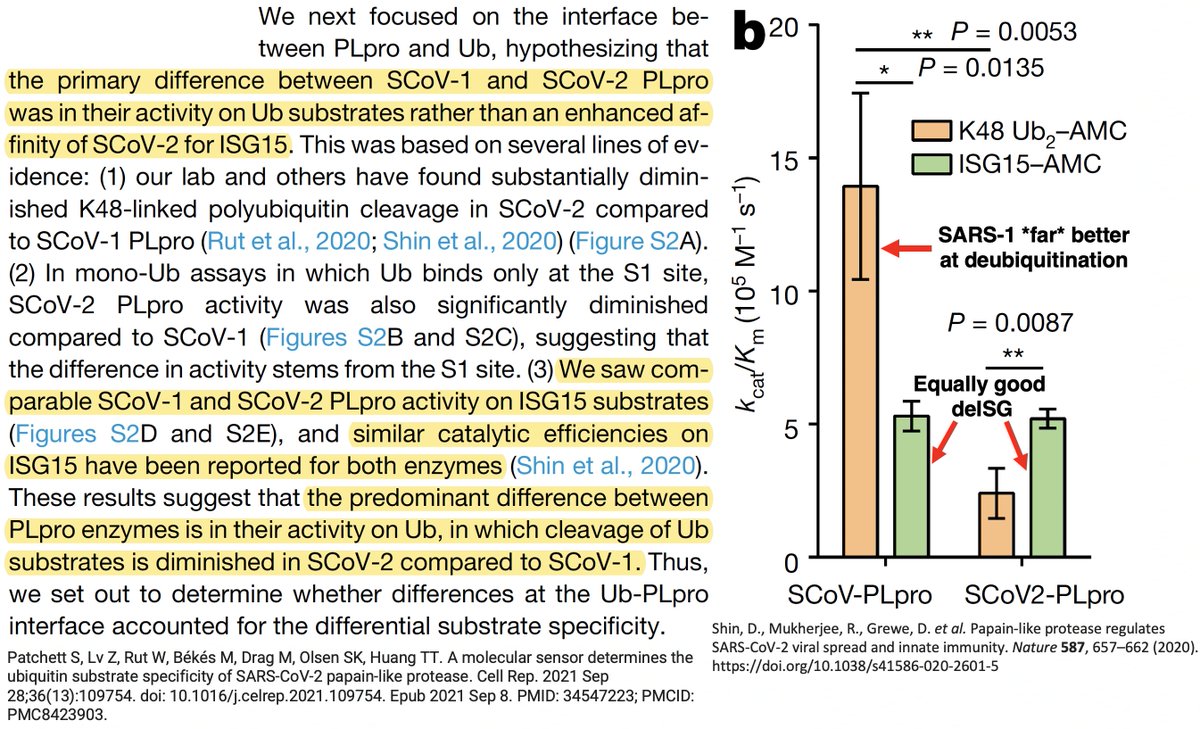
The PLpro-ologists noticed several amino acids that differ between the SARS-1 & SARS-2 PLpros, so they tested out mutant PLpros to see which of these differing AA residues made SARS-1 so much better at DUB than SARS-2. PLpro:232 turned out to be one key. 31/58
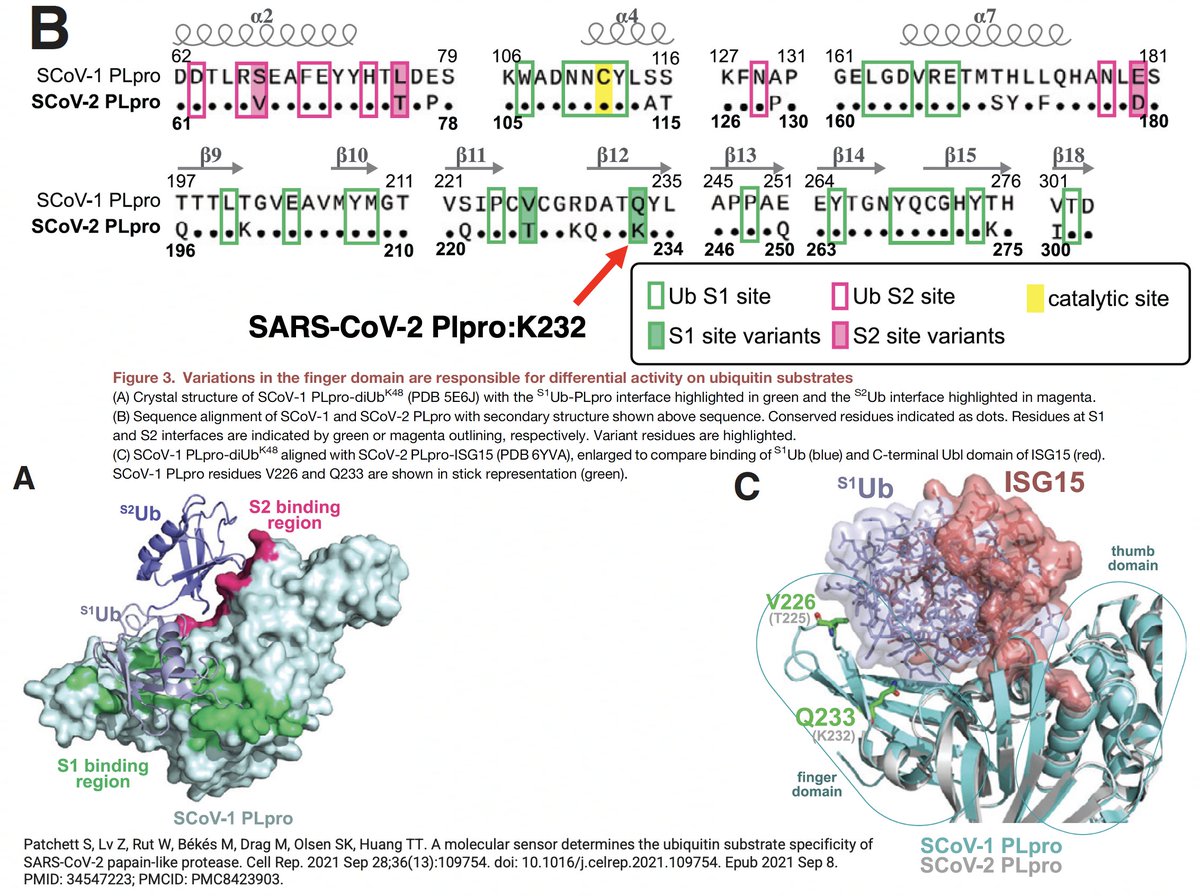
PLpro:232 amino acid is K in SARS-2 but Q in SARS-1. When researchers substituted Q for K in the SARS-2 PLpro (K232Q), its DUB activity increased to SARS-1 levels. The reverse change (PLpro:Q233K), devastated SARS-1's DUB ability, driving it below SARS-2 levels. 32/58
About 2 years after the study came out, @solidevidence, with an assist from @jasnah_kholin, deciphered the PLpro-ologists' obscure terminology and discovered… 33/58
https://twitter.com/wanderer_jasnah/status/1668677961464049665
…that what the PLpro-ologists call PLpro:K232Q everyone else knows as NSP3_K977Q or ORF1a:K1795Q—the same mutation we frequently see in chronic-infection and cryptic WW sequences and rarely anywhere else (except Gamma). 34/58
https://twitter.com/SolidEvidence/status/1668995910141894659
(What bothers me is not that the PLpro experts use a terminology of their own, which is perfectly fine, but that they never—not even parenthetically—say what the corresponding ORF1a or NSP3 residues are. I wasted a ton of time trying to find this.) 35/58
Intriguingly, ORF1a:K1795Q is not only a mutation to the SARS-CoV-1 residue, it's also a reversion to the AA possessed by the bat coronaviruses BANAL-52 and RaTG-13, whose PLpro sequences are 98% similar to that of SARS-CoV-2. Thx to @NitscheLab for alignment. 36/58
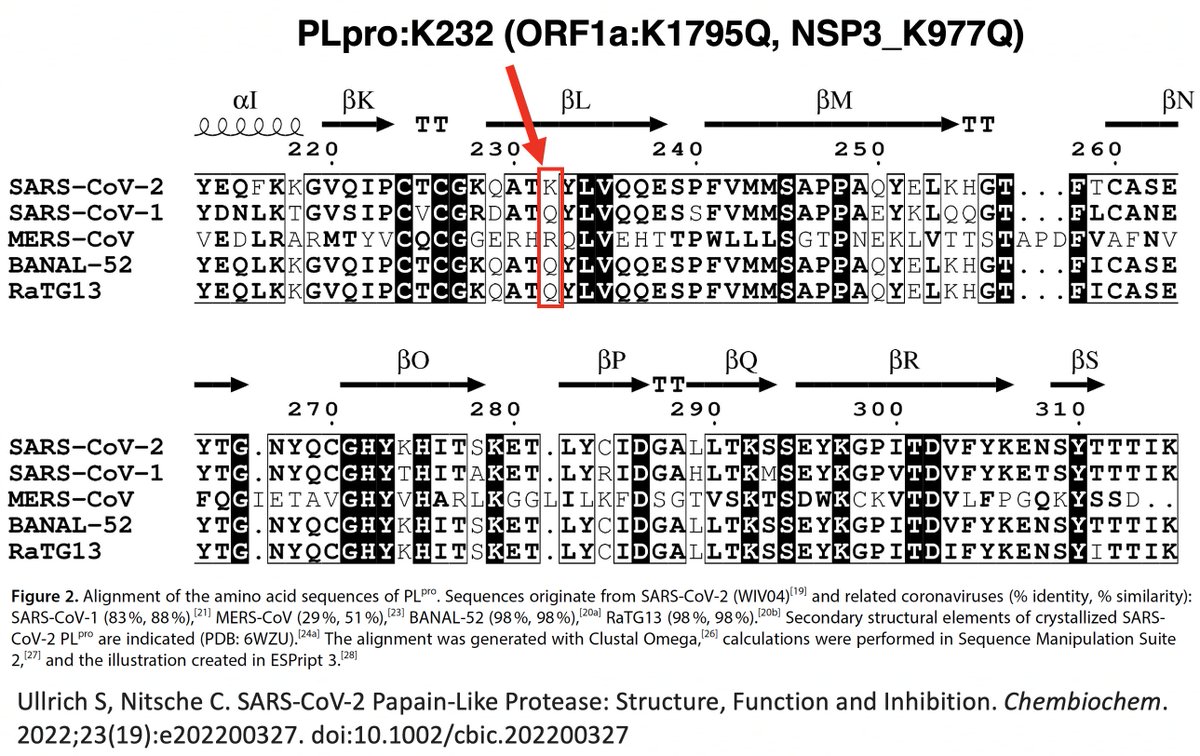
What about the 2nd PLpro mutation I mentioned upthread? That would be ORF1a:T1638I (NSP3_T820I), which @shay_fleishon & @SternLab recently found to be one of the most common mutations in the chronic sequences found by their AI search program. 37/58
https://twitter.com/SternLab/status/1681148509301485571
They noted that, unlike most common mutations seen in chronic-infections, ORF1a:T1638I seems harmful to viral fitness. They also note it is located in a T-cell epitope for one HLA type. This may partly explain its prominence in chronic sequences, but… 38/58

…while reading various studies on PLpro, I discovered that in 2020 a group of researchers—including @CiesekSandra, @Donghyuk_Shin_, & @iDikic2—found, while comparing the SARS-CoV-2 and SARS-CoV-1 PLpro structures… 39/58
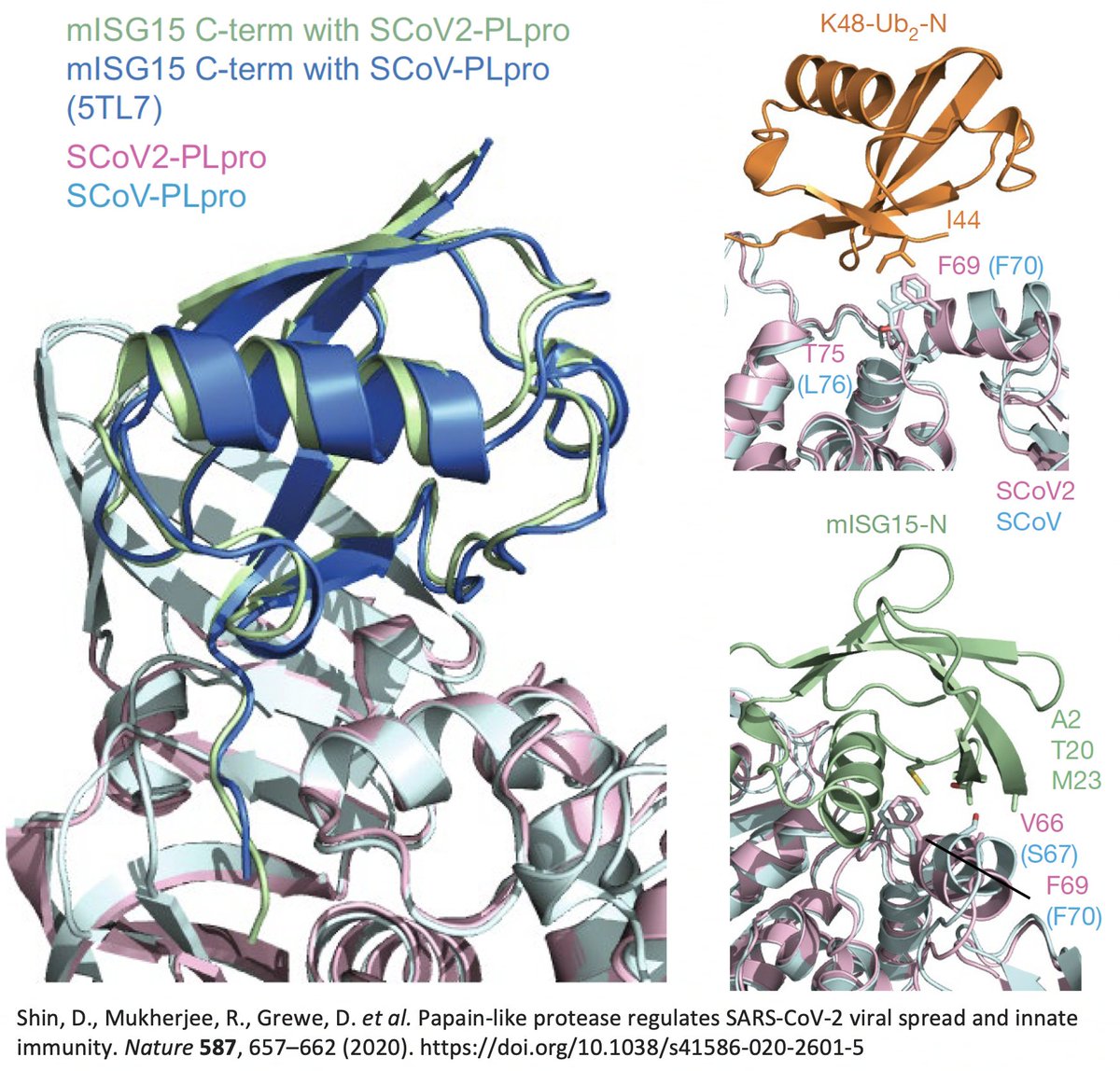
…that "the main difference" between the SARS-CoV-1 & SARS-CoV-2 PLpro structures when bound to ubiquitin was PLpro:T75—better known as ORF1a:T1638 or NSP3_T820. In SARS-CoV-1, the amino acid at this site is leucine—PLpro:L76. 40/58

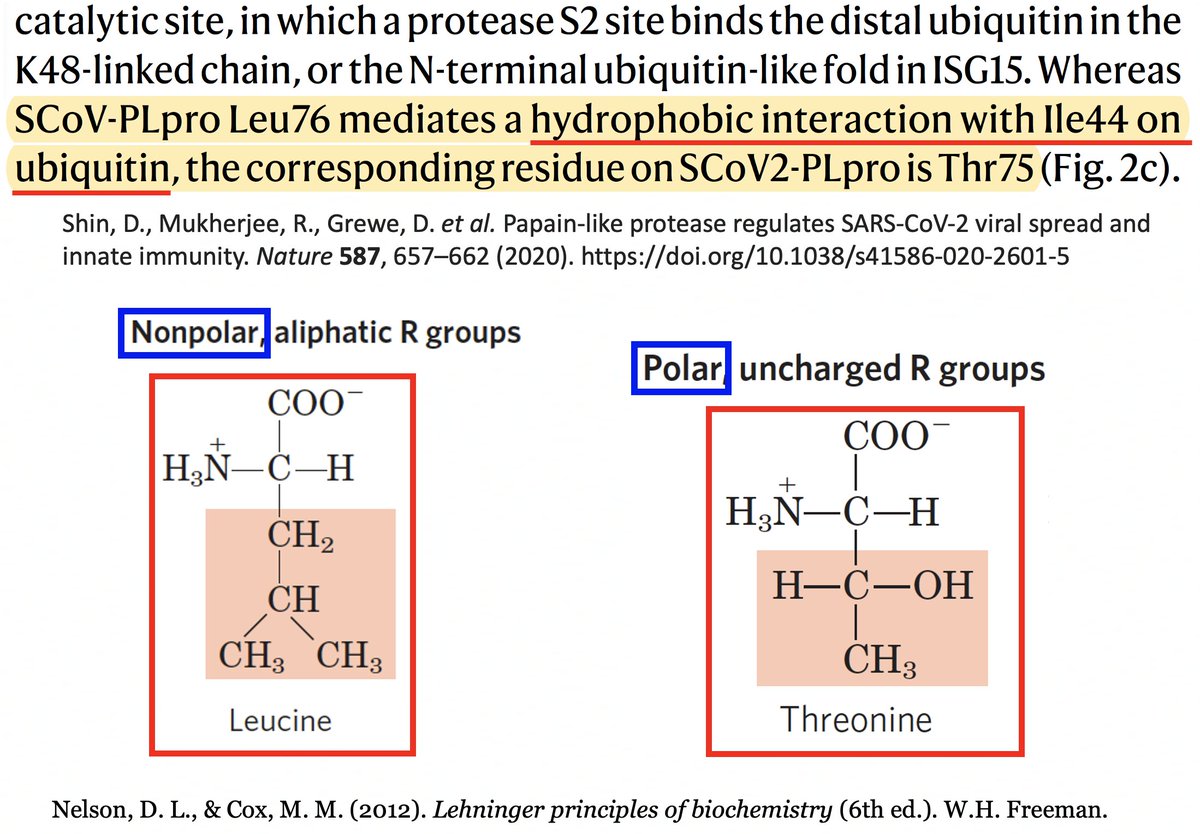
T (threonine) has a polar, or hydrophilic (water-loving) side chain, whereas L's (leucine) is non-polar, or hydrophobic. The authors note that PLpro:L76 in SARS1 creates a bond with a hydrophobic residue (I) in ubiquitin, a bond absent—due to the polar T—in SARS2. 41/58
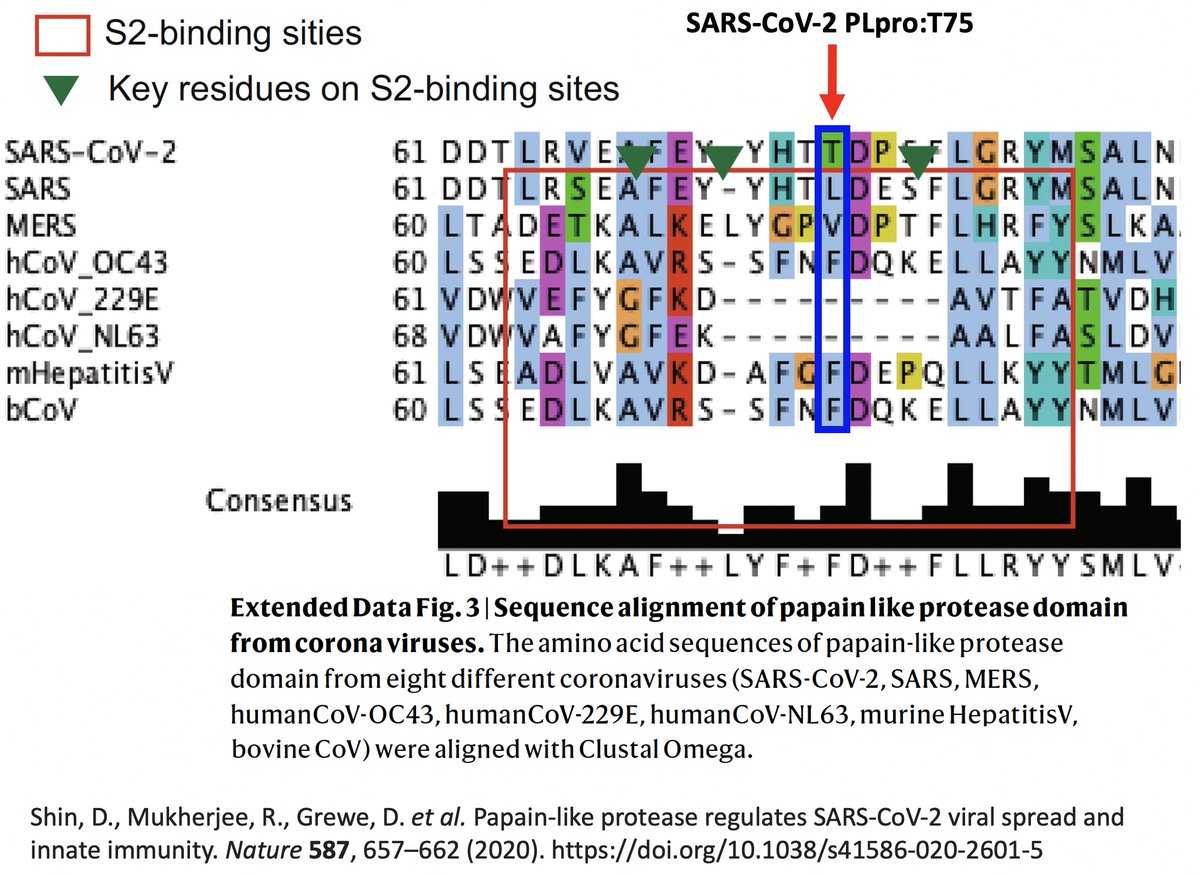
It’s interesting to note that of the eight coronaviruses compared in this study, SARS-CoV-2 is the outlier at this site. Every other CoV has either a hydrophobic/nonpolar residue there—F(3), L(1), V(1)—or a deletion (2). 42/58
So they created two SARS2 PLpro mutants w/hydrophobic AA at this site—PLpro:T75A and PLpro:T75L—then measured their ability to deubiquitinate (DUB) proteins. T75L greatly increase DUB activity (though not to SARS1 levels) without harming its deISG ability. 43/58
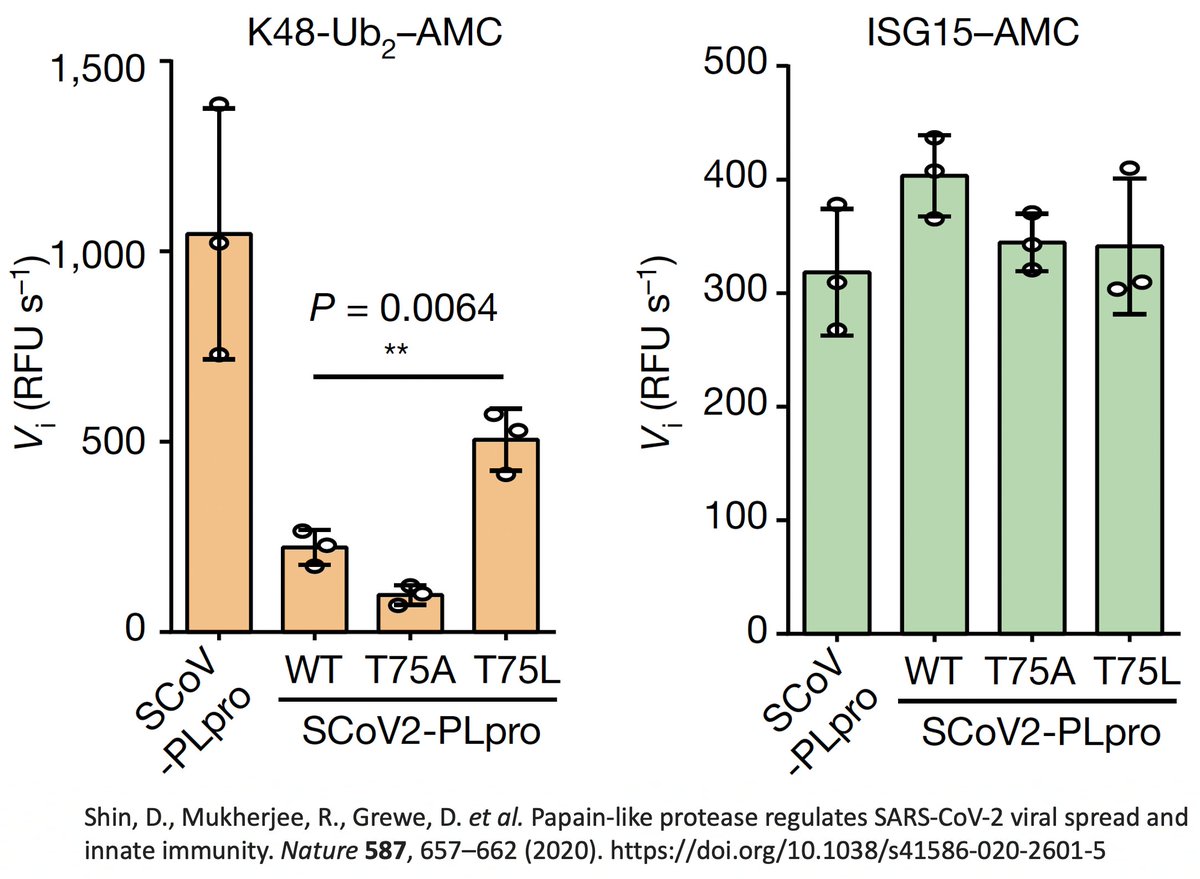
PLpro:T75A (ORF1a:T1638A) actually decreased DUB, likely because of its small side chain. The authors concluded that the large size & hydrophobic nature of the AA at this PLpro site to be "crucial determinants for ubiquitin binding." 44/58

ORF1a:T1638L requires a 2-nucleotide change, which are very rare. However, isoleucine (I) is very similar to L, both in size & hydrophobicity, & only requires a 1-nuc mutation. And by my count, T1638I is the most common non-spike mutation in chronic-infection sequences. 45/58
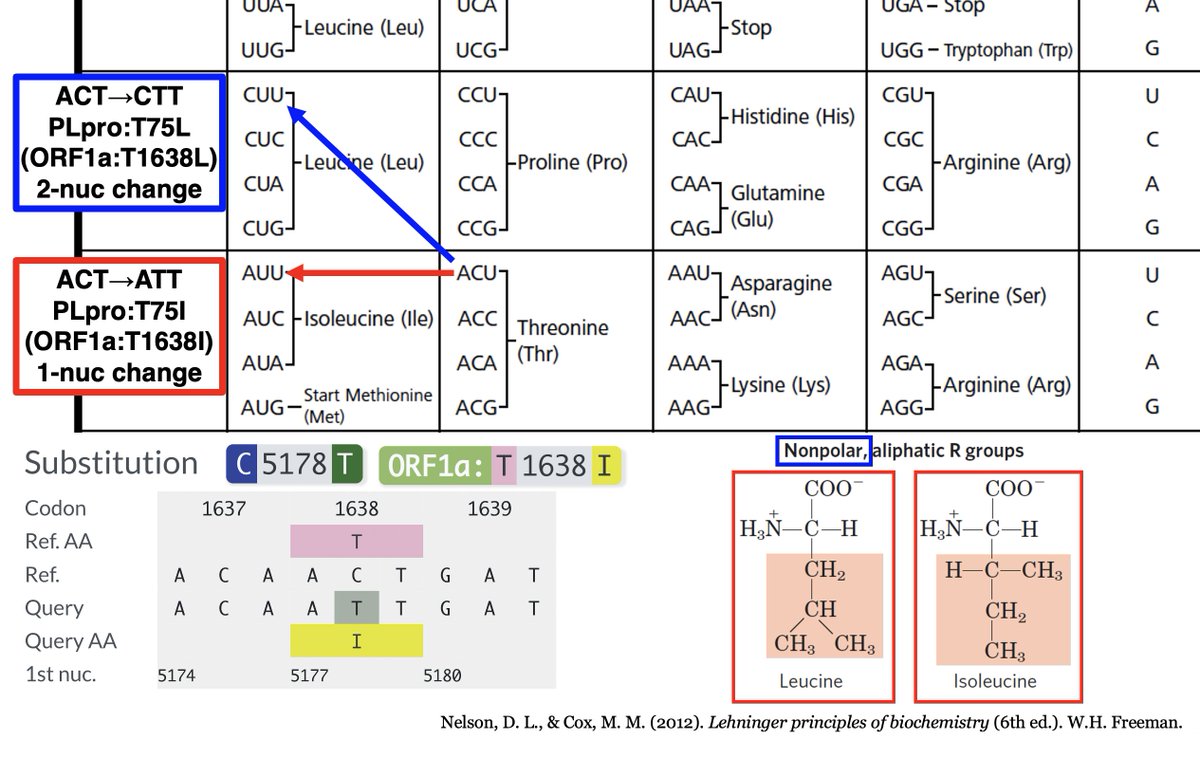
In other words, this mutation had *the exact same effect* as the other extremely common PLpro mutation in chronic infections—ORF1a:K1795Q. Both greatly increase the SARS2 PLpro's ability to deubiquitinate proteins, modulates innate immune activation. 46/58
This shows a remarkable evolutionary convergence in *function* occurring at two different sites in the SARS-CoV-2 genome. It indicates that enhanced deubiquitination is a key element in chronic infections but one that hasn't been selected for in circulating variants. 47/58
But it begs the question: Why is enhanced DUB selected for in chronic infections? And given that this enhanced ability appears to have no negative effect on PLpro's ability to dice up the polyprotein or deISGylate, why is this *not* selected for in circulating variants? 48/58
This is a very difficult question to answer because of the fiendishly complex nature of the innate immune reaction and the promiscuous activity of both ISG15 and ubiquitin. The cascading upstream and downstream reactions are endless. 49/58
Another question: ORF1a:K1795Q was in Gamma, a successful variant famous for causing reinfections & severe illness. To what degree was K1795Q responsible for these aspects of Gamma? (H/t @mike_famulare for hospitalization rates by variant chart) 50/58
https://twitter.com/paredesmig/status/1496594484959666176
Another Gamma peculiarity was revealed by @VirusesImmunity. MHC-I is a cell-membrane surface protein essential to activating cytotoxic (CD8) T cells. Gamma (P.1) was the worst variant of all—worse than WT—at suppressing MHCI transcription. 51/58
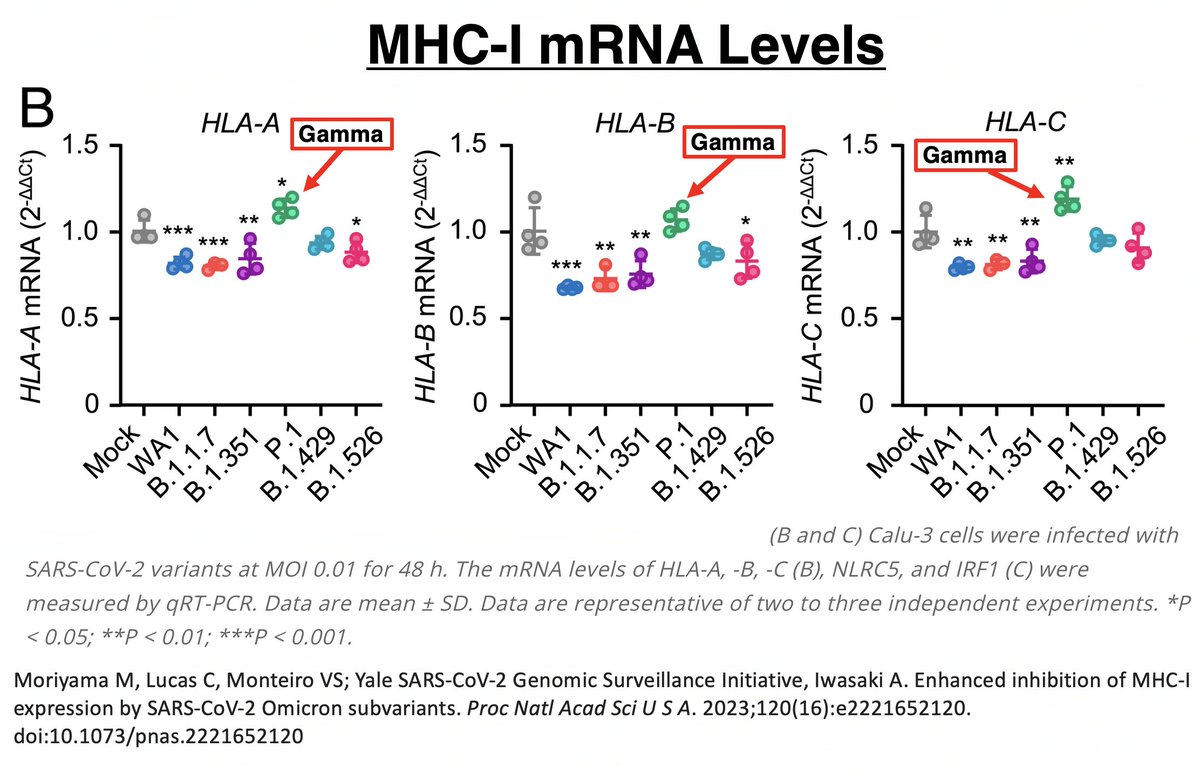
In other words, MHC-I mRNA was being more copiously produced in Gamma than all other variants. Despite this, Gamma had the lowest MHC-I cell surface expression of all. Gamma clearly has a way of impeding surface expression after mRNA transcription. 52/58
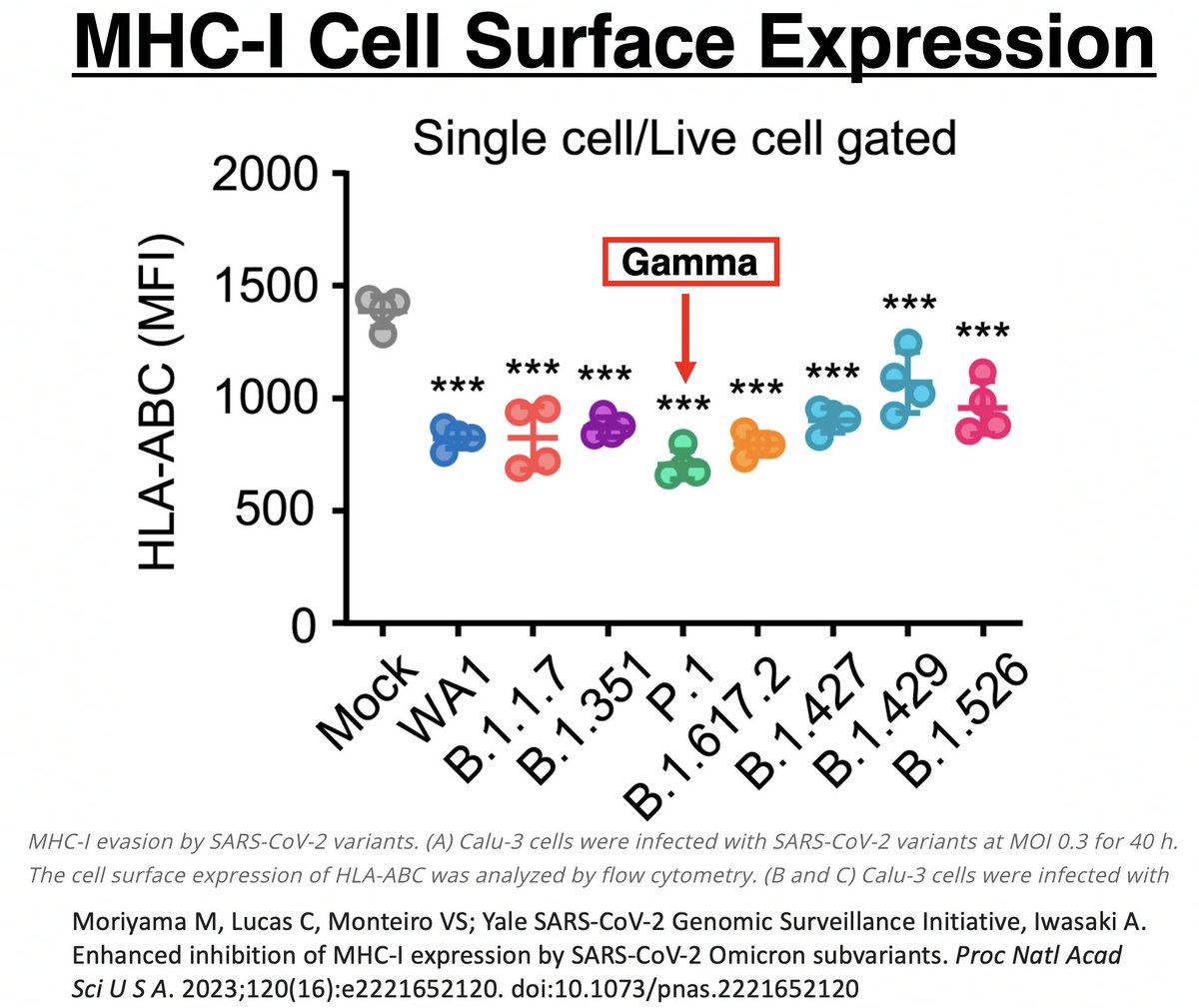
Could it be due to ORF1a:K1795Q (PLpro:K232Q)? If so, could ORF1a:T1638I also sabotage MHC-I cell surface expression? Mutations to evade T-cell responses is exactly what you would expect to see in chronic infections. 53/58

Several other PLpro mutations repeatedly appear in chronic infections—two of them adjacent to T1638I—ORF1a:D1639N & ORF1a:P1640S/L/H. Do these have a similar function as T1638I? Or are they T-cell epitopes, as I’ve long suspected? 54/58
Finally, while reading about ISG15 and ubiquitin, I found that in chronic hepatitis C infections, ISG15 is known to play a proviral role by inhibiting the IFN response. The SARS-CoV-2 PLpro mutations found in chronic infections we've discussed don't affect ISG15. But… 55/58

… to some extent, ubiquitin & ISG15 compete for attachment to proteins. Could the increased DUB activity conferred by these mutations free up locations for ISG15 to attach to, and is it possible ISG15 could have a similar proviral role in chronic Covid? 56/58

This idea is almost certainly wrong, but I'm tossing it into the ether anyway. Could be 1000 things happening here, but I can only come up w/ideas relating to what I've studied recently. I'm like the guy in that joke who lost his keys & only searches under a streetlight. 57/58

But I never really got that joke. That guy always seemed sensible to me. Searching in complete darkness is hopeless. Searching in a small area that has light isn't much better, but at least there's a tiny chance of success. And that's usually the best we got. 58/58
• • •
Missing some Tweet in this thread? You can try to
force a refresh